According to malaria statistics, two hundred and nineteen million cases and four lakh and thirty-five thousand deaths were accounted for in 2018 (World Health Organization, 2018). The finite number of drugs and loss of effectiveness of available drugs against disease is the principal cause of the expansion of morbidity and mortality. The drifting of interest towards the exploration of new drugs and their targets is necessitated. The emergence of drug resistance along with multidrug-resistant cases is appending to the complexity of the disease. Among the pathogenic species, Plasmodium falciparum accounts for the notable worldwide infections. It belongs to the phylum Apicomplexa, a group of protozoa containing multi-membrane surrounded organelle known as apicoplast. The discovery and indispensable nature (He et al., 2001) of apicoplast have unfolded new opportunities in the area of drug discovery (Ralph et al., 2004).
Apicoplast houses four major metabolic processes, but the discovery of various scavenging mechanisms for fatty acids and heme in the erythrocytic stage contravened their drug target ability (Nagaraj et al., 2013; Vaughan et al., 2009). In Plasmodium, the desideratum of isoprenoid and its precursors are met by an alternative pathway known as the 2-C-methyl-D erythritol-4-phosphate (MEP) pathway (Rohmer and Rohmer, 1999). The enzymes participating in the pathway are transported to the apicoplast from the cytoplasm via signals present at N-terminus (van Dooren and Striepen, 2013). The multifarious isoprenoids are synthesized from their precursor subunits such as Isopentenyl Pyrophosphate (IPP) and Dimethylallyl Pyrophosphate (DMAPP). The diversity of isoprenoids contributes to innumerable roles such as prenylation and glycosylation of proteins, regulation of gene expression, constituents of membrane and electron transport chain (Hunter, 2007; Sacchettini and Poulter, 1997), quinone production, vitamin E and carotenoid biosynthesis (van der Meer and Hirsch, 2012).
The study reported chemical rescue of isopentenyl pyrophosphates auxotroph which are entirely dependent on exogenous IPP, defines IPP synthetic pathway is the sole function of apicoplast in the blood stage (Cassera et al., 2004; Yeh and DeRisi, 2011). The inevitable role of the MEP pathway in both hepatic and gametocytogenesis is also reported (Wiley et al., 2015). The absence of any scavenging mechanism of isoprene units in Plasmodium and the requisite in erythrocytic stages acclaims this pathway as a sole drug target for antimalarial research. Gene knockout studies in bacteria explained the essentiality of all enzymes of the pathway (Missinou et al., 2002). The disparity concerning humans diminishes the target toxicity issues and provides numerous protein targets which can be harnessed further for inhibition and drug research.
The target identification performed through a systemic inhibition study (Singh and Ghosh, 2013) categorized the IPP pathway enzymes into two groups. The first category is responsible for flux reduction while inhibition of another set of enzymes causes a significant accumulation of their substrates which creates instability in the cell. In this facet, 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXP Reductoisomerase/DXR) or IspC (EC1.1.1.267), the enzyme against which several studies of inhibition have been reported (Borrmann et al., 2006; Takahashi et al., 1998) belong to the first category. The enzyme converts 1-deoxy-D-xylulose-5-phosphate (DOXP) to MEP (Takahashi et al., 1998). The enzyme has a homodimer structure comprising of N-terminal, catalytic, and C-terminal domains and an additional linker region that connects the C-terminal and catalytic domains. The active site present in the catalytic domain remains in the open conformation and accommodates its substrate which is converted into closed conformation to perform catalytic function (mac Sweeney et al., 2005). The potent inhibition of the enzyme is observed by the use of a natural antibiotic, fosmidomycin, and its derivatives (Jomaa et al., 1999; Steinbacher et al., 2003). PfDXR inhibition is also reported to be an effective strategy to treat uncomplicated malaria in clinical phase II trials using fosmidomycin alone or in combination with clindamycin (Borrmann et al., 2006). However, due to the recrudescence of the disease, further alterations in fosmidomycin or another compound are required which may lead to the curing of the disease.
The benzodiazepine (BDZs) scaffold is found in many compounds which are active against several protein targets (Evans et al., 1988; James et al., 1993; Nicolaou et al., 2000; Schneider and Schneider, 2017). BDZs belong to a conventional class of drugs exhibiting good oral bioavailability (Rotonda et al., 1996). Also, the scaffold is contained in several compounds having inhibitory activity against ɣ-secretase (Churcher et al., 2003) and farnesyl protein transferase (Hunt et al., 2000; James et al., 1993). Some BDZs derivatives show antitumor (Dourlat et al., 2007) and antimalarial properties (Ettari et al., 2008; Micale et al., 2006).
Owing to the importance of the benzodiazepine structural and biological features (Fig. 1), our study is inclined to the development of a series of benzodiazepine analogues on the basis of its conventional scaffold. Several Lewis acid/Metal catalysts are used in the synthesis of these N-heterocyclic compounds (Ghorbani-Vaghei and Veisi, 2010; Ha et al., 2010; Jacob et al., 2011; Neochoritis et al., 2010; Radatz et al., 2011). Most of the strategies are involved in either multistep synthesis or special conditions such as anhydrous conditions, use of expensive reagents, drastic reaction conditions, the formation of various side products, longer reaction time, poor yields, and use of toxic solvents (Bennamane et al., 2008; Päsha and Jayäshankara, 2007). Given these drawbacks associated with some of the synthetic procedures, herein we presented a novel synthetic strategy for the diversity-oriented synthesis of benzodiazepines employing environmentally benign Ca(II) as the green catalyst (Pareek et al., 2016; Yaragorla et al., 2016a). The study reports the synthesis of benzodiazepine analogues, their antimalarial, and enzymatic features which purveys them to be a new class of DXR inhibitors (Fig. 1).

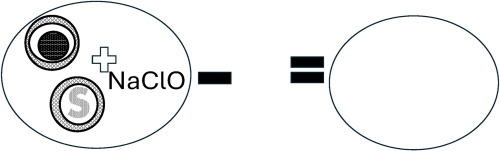


留言 (0)