
記住我










Retinitis pigmentosa (RP; OMIM:268000) is a prevailing form of inherited retinal dystrophy (IRD) and a major cause of blindness worldwide. RP is inherited in autosomal dominant, autosomal recessive or X-linked inheritance patterns following Mendel’s law of inheritance, with very few exceptions.1 So far, more than 80 genes have been reported as genetic causes of RP. As with other Mendelian disorders,2 3 40%~70% of patients with RP do not have a molecular diagnosis.1 4–6 Nevertheless, advances in sequencing technology have enabled scientists to reveal the genetic causes of RP.1 6–9 Previous studies have demonstrated that EYS, USH2A, RPGR and RHO are the frequent causative genes of RP across ethnicities.1 6 7 For Japanese patients with RP, EYS, which is the cause of autosomal recessive RP (ARRP), has been reported as the most frequent causative gene.6 10
We previously reported the targeted short-read sequencing of 83 RP-associated genes in 1204 Japanese patients with RP .11 In that study, custom-made multiplex PCR-based targeted resequencing was performed using a next-generation sequencer. As a result, causative genes were determined in 29.6% of patients; however, the remaining cases remained unsolved. Importantly, the study indicated that more than a quarter of unsolved patients had one pathogenic variant in EYS,11 suggesting that EYS is a promising candidate as the causative gene.11 Although the exact reason for this observation has not been clarified, it is possible that variants that are difficult to identify by short-read targeted resequencing, such as structural variants (SVs) and intronic variants, are another causative variant. Indeed, SVs, intronic variants and hypomorphic variants have been uncovered as a second pathogenic variant in patients with IRD.9 12–15 Therefore, we considered whether carriers of pathogenic variants in EYS have second causative variants that are not detectable by short-read target sequencing.11
Long-read sequencing technology has several advantages compared with short-read sequencing, such as accurate SV detection.16 17 In order to increase the number of solved patients with RP and to deepen our knowledge about the genetic basis of RP, here we conducted a sequencing study of 15 patients with RP who are carriers of one pathogenic variant in EYS using long-read sequencing and examined the frequencies of the identified SVs from an independent patient group in our previous study (n=1189).11
Materials and methodsStudy patientsThe clinical diagnosis of RP was based on the patient’s history, visual field and electroretinography outcomes, as well as ophthalmological findings by trained ophthalmologists. We selected 15 cases (figure 1) with heterozygous pathogenic variants in EYS from our previous study11 for whole-genome long-read sequencing. To assess founder effects of the identified variants in the Japanese population, we examined the frequencies of the SVs in the independent patient group from our previous study (n=1189).11 For validation purposes, we performed multiplex PCR-based targeted sequencing18 using the independent RP patient set.
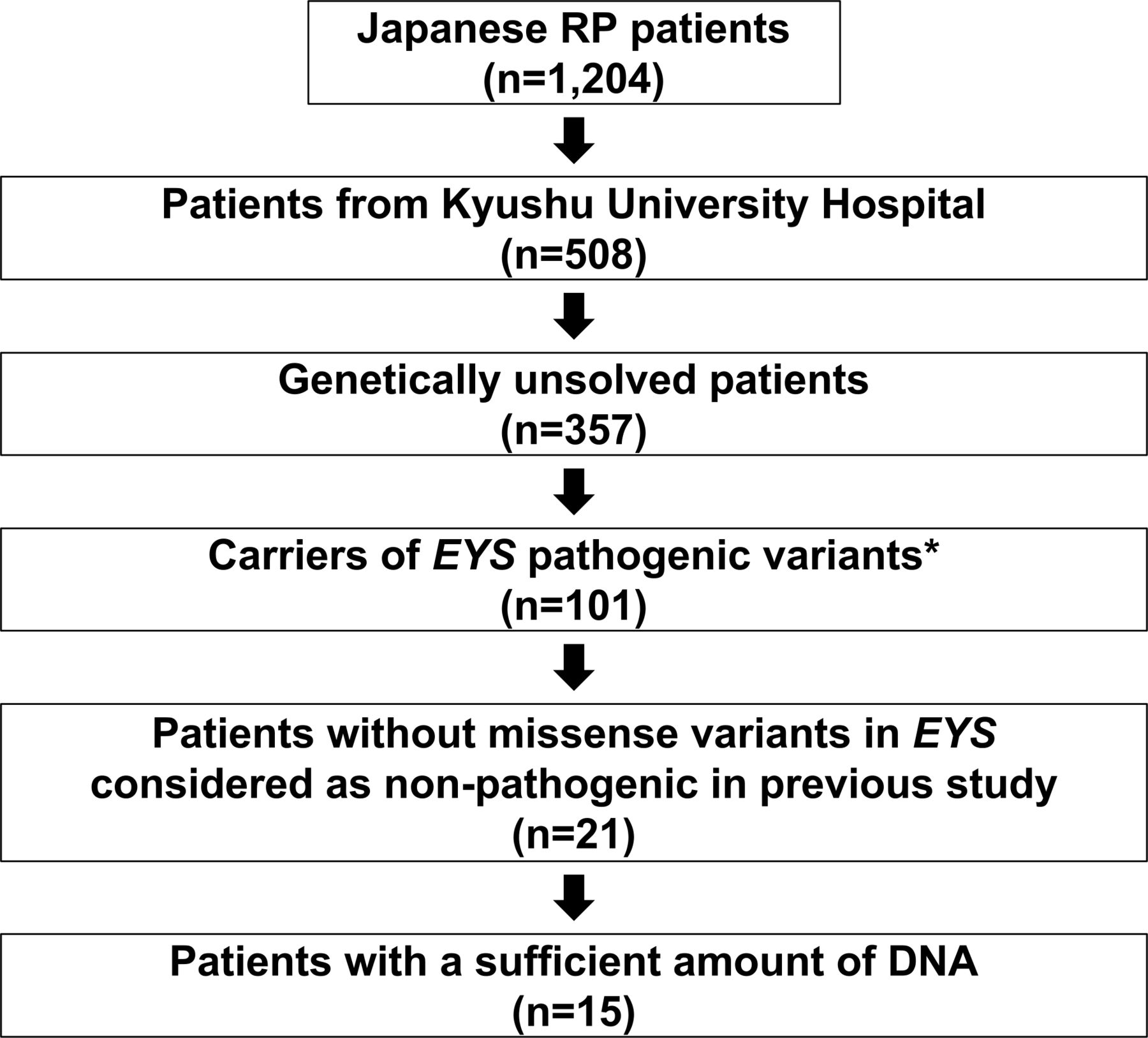 Figure 1
Figure 1 Details of the sample selection. Of the 1204 cases included in the previous study, 508 were patients with retinitis pigmentosa (RP) at Kyushu University Hospital. Among them, we selected 101 genetically unresolved cases with heterozygous pathogenic variants (known pathogenic or loss of function variants) in the EYS gene. Among these cases, 80 cases had missense variants of unknown pathogenic significance in addition to the above-mentioned pathogenic variants. Considering the possible pathogenesis of missense variants not considered pathogenic in our previous study, patients with such variants were excluded. Of the remaining 21 cases, 15 cases with a sufficient amount of DNA were selected for long-read sequencing using the Oxford Nanopore sequencer.
Library preparation and sequencing for whole-genome long-read sequencingLibraries for long-read sequencing were prepared using the SQK-LSK109 Ligation Sequencing Kit (Oxford Nanopore Technologies) following the manufacturer’s protocol, and the sequencing itself was performed according to the manufacturer’s protocol using FLO-MIN106 R9.4.1 flow cells (Oxford Nanopore Technologies) for 96 hours. The number of sequencing runs was adjusted to obtain at least 45 Gb of data for each sample (online supplemental table S1).
Identification and validation of SVs in RP-associated genesBase-calling was performed using Guppy V.4.4.1 (Oxford Nanopore Technologies). We mapped reads to the reference sequence (GRCh38) and focused on SVs, because long-read sequencing does not have high detection accuracy for single-nucleotide variants (SNVs) or short indels.17 19 We used the CAMPHOR17 pipeline to detect indels (≥50 bp), inversions, duplications and translocations. After the variant-calling, SVs longer than 1 Mb were excluded due to the possibility of variant-calling errors.
To select candidate pathogenic SVs for RP, we analysed EYS and another 88 RP-associated genes listed in the Retinal Information Network (RetNet) (https://sph.uth.edu/retnet/) as of 14 April 2021 (online supplemental table S2). We proceeded to search for the following types of SVs: (1) SVs in the coding region of EYS, which is one of the most promising candidate causative genes, (2) SVs which overlap human retina-specific exons of EYS that may play important roles in retinal diseases,20 (3) SVs within 500 bp of the exon boundaries of EYS that could affect splicing or promoter functions and (4) SVs in the RefSeq coding regions of other RP genes that could be causative variants. Variant classification was performed for the detected variants according to the guidelines of the American College of Medical Genetics and Genomics (ACMG).21
To prioritise pathogenic SVs, the allele frequencies (AFs) of the SVs were compared with those in our in-house long-read whole-genome sequencing data (53 Japanese healthy control samples), Genome Aggregation Database (gnomAD SVs v2.1) and dbVar database.22 We removed variants using the following criteria: (1) variants found in the 53 Japanese control samples, and (2) variants with an AF>0.5% for recessive genes and >0.01% for dominant genes in gnomAD SVs or dbVar.
Among the SVs overlapping the EYS region, we excluded three large SVs (110 Mb inversion, 82 Mb inversion and 97 Mb translocation) to avoid variant-calling errors using the following criteria: (1) the size of the SVs was too large, (2) there were only two reads supporting the breakpoints of the SVs and (3) the 1 kb sequence around the breakpoint was aligned with multiple regions of the human genome and likely to be a repeat region. We noted that the excluded SVs were less likely to be pathogenic, since their breakpoints did not overlap with the regions of RP-associated genes.
We tried to identify SNVs from long-read data and compared the results with our previous study11 (online supplemental table S3). Long-read sequencing identified a much larger number of SNVs than short-read sequencing, suggesting a higher error rate in the former. Therefore, we did not take SNVs into consideration in the current study.
To validate the identified SVs, we amplified the SV junction regions by PCR using KOD Multi&Epi enzyme (TOYOBO), and amplicons were subjected to Sanger sequencing.
To examine the frequencies of the SVs in the independent patient group (n=1189),11 we performed multiplex PCR-based targeted sequencing. Three types of PCR primer sets were designed for each of the identified SVs to amplify the upstream breakpoint regions, downstream breakpoint regions and deleted regions of the large deletions (online supplemental figure 1 and table S4). The details of the sequencing method have been described previously.18
ResultsWe selected 15 patients with RP with one pathogenic variant in EYS from our previous study11 and confirmed that all of them had typical RP. We performed whole-genome sequencing with the Oxford Nanopore long-read sequencer. The average number of reads and their lengths were 6 623 354.7 and 8136.7 bp, respectively (online supplemental figure 2, table S1), and 93.7% of the reads were aligned to the human reference genome (GRCh38) (online supplemental table S5). The mean depth of the EYS region was 16.5. The most the EYS region (94.1%) was covered by ≥10 reads (online supplemental figure 3, table S6).
We confirmed that the pathogenic variants of EYS detected in our previous targeted resequencing study11 could be found by long-read sequencing all patients (online supplemental figure 4, table S7). We then focused on identified SVs. In the 15 patients with RP sequenced, 46 899 SVs consisting of 22 786 deletions, 22 790 insertions, 89 inversions, 765 duplications and 469 translocations were identified across the genome. We observed 176 exon-overlapping SVs per patient on average, which included 103 deletions, 44 insertions, 7 inversions, 10 duplications and 12 translocations (online supplemental table S8). Next, we focused on SVs overlapping 89 RP-associated genes and found 15 candidate SVs (table 1). In the ARRP genes, 12 candidate SVs in EYS (9 deletions, 3 insertions), 1 deletion and 1 insertion in RP1L1, and 1 duplication in ARHGEF18 were identified. The three deletions and two insertions in EYS did not overlap with the coding region but were within 500 bp of an exon (table 1). However, no exon-overlapping SVs were detected in autosomal dominant and X-linked recessive RP genes.
Table 1Summary of SVs identified in RP-associated genes
Two large exon-overlapping deletions in EYS were found in OPH641 and OPH861 (figure 2A). The lengths of the identified deletions were approximately 376 kb and 395 kb in OPH641 and OPH861, respectively. We performed Sanger sequencing and detected the exact breakpoint sequences for each SV. One deletion which involved six exons (exon 23–28) was accompanied by a 4 bp insertion within the breakpoints (NM_001142800.1:c.3443+14 421_5927+13 006delinsTCAT; figure 2A). This deleted region fully encompasses and overlaps a region in which an inverted duplication was previously reported.23 Similarly, a 6 bp insertion within the breakpoints was found in the other deletion that overlapped with six exons (exon 30–35) of EYS (NM_001142800.1:c.6079–30740_7055+41 631delinsCATAAT; figure 2A). The breakpoints of both SVs were located in repetitive sequences (figure 2B). Insertions of short fragments were detected at the breakpoints, which suggests that the insertions were caused by fork stalling and template switching (FoSTeS)/microhomology-mediated break-induced replication (MMBIR).24 According to the ACMG guidelines,21 the PVS1 and PM2 criteria were applied to these SVs, and they were considered likely pathogenic. We also confirmed the pathogenicity of SNVs identified in our previous study (Supplementary Note). Considering that two causative variants were present in the ARRP causative gene, we regarded EYS as the causative gene for both OPH641 and OPH861. Clinical information for these two patients is provided in the Supplemental material (online supplemental figure 5). Both probands were sporadic cases according to the medical interviews. Although we were unable to investigate the segregation of the SVs with phenotypes in the pedigrees, this family history is consistent with the ARRP gene (EYS) being the causative gene. For the other thirteen candidate SVs for the ARRP gene, three control data (in-house, gnomAD SVs, dbVar) were used to prioritise variants by AF. A 683 bp heterozygous deletion in RP1L1 found in OPH690 remained, and the other 12 candidates were excluded. According to the ACMG guidelines,21 the PVS1 and PM2 criteria were applied to this SV, and it was considered likely pathogenic. However, the other pathogenic variants in RP1L1 were not detected in OPH690 by our previous targeted resequencing (online supplemental table S9),11 therefore, we did not consider RP1L1 as the causative gene for this patient.
 Figure 2
Figure 2 The causative structural variants (SVs) in EYS. (A) A schematic diagram of heterozygous deletions (top), an integrative genomics viewer (IGV) visualisation of the breakpoints of deletions (middle), and the exact breakpoint sequences confirmed by Sanger sequencing (bottom) in OPH641/OPH861 are shown. In each patient, 4 bp (OPH641)/6 bp (OPH861) insertions were identified within the deletion breakpoints. For example, exon. (B) The genetic elements to which the breakpoints belong to are shown. The downstream breakpoint of the deletion in OPH861 belongs in a repetitive region, and others belong in Long Interspersed Elements (LINE). Red and blue bars display the downstream/upstream breakpoint junctions. The dashed rectangles indicate the genetic elements where the breakpoints are localised.
Finally, we examined the frequencies of the two large SVs in EYS in other patients with RP. We conducted multiplex PCR-based targeted sequencing18 to evaluate these SVs in 1189 independent Japanese patients with RP. However, the two SVs were not detected, suggesting that they are not founder mutations.
DiscussionIn this study, we identified likely pathogenic SVs in two previously unsolved RP cases by long-read sequencing. The proportion of patients with a new molecular diagnosis was 2 out of 15, which is consistent with previous reports using short-read whole-genome sequencing (4.8%–12.5%).23 25
Short-read and long-read sequencing can be applied to whole-genome sequencing. Although the former has the advantage of detecting SNVs and short indels, functional experiments are generally required to assess the causality of intronic variants. On the other hand, long-read sequencing may identify pathogenic SVs, such as exon-overlapping SVs. Indeed, our study identified two likely pathogenic SVs. Although the utility of long-read sequencing for Mendelian diseases is not well established, this study indicates that long-read sequencing contributes to the molecular diagnosis of Mendelian diseases. Our previous and current studies have comprehensively investigated SNVs and short indels in the coding regions and SVs in EYS for 15 cases.11 However, 87% (13/15) of the cases still remain unsolved. Considering that variants in coding regions were well investigated with sufficient coverage (online supplemental table S6), variants in non-coding regions could be the cause for these patients.
This study did not analyse intronic SNVs for three reasons: (1) pathogenic deep intronic EYS variants have not been reported previously, (2) the pathogenicity of intronic variants requires functional validation and (3) long-reads sequencing does not have sufficient accuracy for SNV identification. While long-read sequencing has contributed to molecular diagnosis in some cases, an inadequate search for variants in deep introns may result in the low detection rate of pathogenic variants. Therefore, further evaluation of non-coding regions in patients with unresolved RP by short-read sequencing and functional validation should increase the number of genetically solved patients. Additionally, an analysis of other candidate genes is recommended.
Another limitation of this study is that the haplotypes of the pathogenic variants in the patients could not be examined. A segregation analysis of the patients observed in the present study will further clarify the impact of the identified SVs.
In conclusion, we identified likely pathogenic SVs in an ARRP gene by long-read sequencing. Our results imply that searching for SVs and the comprehensive evaluation of non-coding regions in genetically unsolved cases will contribute to the molecular diagnosis of RP.
Ethics statementsPatient consent for publicationEthics approvalThe ethics committees of Kyushu University Hospital, the University of Tokyo, and all collaborating hospitals have approved this study. All participants have provided written consent to participate in the study. This study was conducted following the principles of the Declaration of Helsinki.
AcknowledgmentsThe super-computing resource was provided by the Human Genome Centre, Institute of Medical Science, the University of Tokyo.
留言 (0)