
記住我
Fast, high quality volumetric imaging facilities many attractive applications in the biomedical studies, such as the large-scale neural activity imaging (Demas et al., 2021; Zhang et al., 2021c), hemodynamic imaging (Wagner et al., 2019; Wagner et al., 2021), neurovascular coupling imaging (Park et al., 2017; Fan et al., 2020), and the cytology research (Hua et al., 2021; Wu et al., 2021), etc. The urgent need for high-speed volumetric imaging has spawned the development of various techniques, such as the multiphoton fluorescence microscopy (Zipfel et al., 2003), the confocal microscopy (Jonkman et al., 2020), and the light sheet microscopy (Bouchard et al., 2015; Voleti et al., 2019), etc. These methods have shown excellent performance and have been commercialized and have advanced biomedical research for years. However, the imaging speed of conventional volumetric imaging microscopy is limited either by its mechanical scanning speed or the detector response speed (Jin et al., 2020; Chang et al., 2021). This makes them typically only possible to image several planes with specific depths at lower speed. Besides, these methods usually rely on complex systems. For example, two-photon microscopy requires expensive lasers and complex scanning systems, and light sheet microscopy requires multiple objectives, etc.
On the contrary, the recently developed light field microscopy (LFM) has shown great advantages in fast volumetric imaging (Levoy et al., 2006; Broxton et al., 2013; Prevedel et al., 2014; Wagner et al., 2021; Wu et al., 2021). Its super volumetric imaging capability has been used in a variety of applications (Prevedel et al., 2014; Pégard et al., 2016; Li et al., 2019; Chen et al., 2020; Sims et al., 2020; Xiong et al., 2021; Zhang et al., 2021a,b), especially in neuroscience research in vivo. In recent years, LFM has developed rapidly. Among the various developments of LFM (Levoy et al., 2006; Nobauer et al., 2017; Guo et al., 2019; Li et al., 2019; Stefanoiu et al., 2019; Cai et al., 2020; Chen et al., 2020; Wagner et al., 2021; Wang D. et al., 2021; Wang Z. et al., 2021; Wu et al., 2021), the Fourier light field microscopy (FLFM) (Llavador et al., 2016; Cong et al., 2017; Guo et al., 2019; Yanny et al., 2020; Yoon et al., 2020; Hua et al., 2021) has unique advantages. By performing the multi-views imaging simultaneously, FLFM can reconstruct a volumetric image with a single exposure, pushing the volumetric imaging speed up to the limit of the camera. More importantly, FLFM takes advantage of its uniform point spread function (PSF) (Cong et al., 2017; Guo et al., 2019), which helps FLFM to avoid the severe reconstruction artifacts of LFM near the focal plane (Broxton et al., 2013; Wagner et al., 2021). Benefiting from the advantages, FLFM has been widely used in many applications, achieving large field-of-view (FOV) (Xue et al., 2020) and depth-of-field (DOF) (Cong et al., 2017; Yoon et al., 2020) with a relatively high resolution (Hua et al., 2021; Liu and Jia, 2021).
However, in the in vivo imaging applications, the FLFM still suffers from strong out-of-focus signals and tissue scattering (Zhang et al., 2021a,c; Zhai et al., 2022). The former is due to the inherent wide-field illumination methods, while the latter is caused by the non-uniform distribution of tissues. These issues not only deteriorate imaging quality, but also increase the burden of reconstruction (Yoon et al., 2020). So far, various techniques have been proposed. Inspired by previous optical sectioning techniques, researchers have integrated the selective-volume-illumination methods [such as confocal illumination (Zhang et al., 2021c), two-photon excitation (Madaan et al., 2021), light-sheet illumination (Wang et al., 2019; Wang Z. et al., 2021), structured illumination (Taylor et al., 2018; Fu et al., 2021), etc.] and the selective-volume-detection methods [such as the computational methods (Zhang et al., 2021a), confocal slit detection (Zhang et al., 2021c), etc.] into the FLFM. These methods have successfully improved the image quality of LFM. However, the former fails in avoiding the tissue scattering physically, and the latter methods require prior assumptions of samples or complex system design.
To overcome the shortcomings of above methods, we have recently proposed the robust Fourier light field microscopy (RFLFM) (Zhai et al., 2022). By introducing the “HiLo” structured illumination and computational reconstruction (Lim et al., 2008; Santos et al., 2009; Lim et al., 2011), we can remove the background signals and the tissue scattered light simultaneously by post-processing. Moreover, it is worth noting that the RFLFM can be easily adopted by adding a deformable mirror device (DMD) in the illumination path of FLFM. However, the main disadvantage of RFLFM is the a-half loss of imaging speed. Same as the optical-sectioning wide field microscopy (Lim et al., 2008; Santos et al., 2009; Lim et al., 2011; Shi et al., 2019), RFLFM needs to take a structured illumination (SI) image and a uniform illumination (UI) image together to recover an optical-sectioning image. This, unfortunately, decreases the imaging speed and increases the storage burden.
Another computed tomography-based microscopy, the structured illumination microscopy (SIM) (Bozinovic et al., 2008; Mertz, 2011; Hagen et al., 2012; Dongli et al., 2013; Zhou et al., 2015), is also widely used. SIM recovers an optical-sectioning image by sequentially changing the phase of the structured illumination patterns with 2π3. Therefore, the traditional SIM needs to take three consecutive images to recover a single optical sectioned image, which is even slower than HiLo. However, inspired from the periodicity of the sine function, SIM has the potential to restore the original imaging speed (Shi et al., 2021). When we switch the SIM illumination patterns continually, the phases of all patterns are physically continuous too. Thus, we can extract new structured illumination periods from the original periods to make up the speed loss.
Here, we propose the structured-illumination and interleaved-reconstruction based Fourier light field microscopy (SI-FLFM). By using SI-FLFM, we can eliminate the background fluorescence in Fourier light field imaging without decreasing imaging speed. We demonstrate the superiority of our SI-FLFM in high-speed, background-inhibited volumetric imaging of various biodynamics in both larva zebrafish and mice in vivo. The results show that our system has achieved great improvements in both imaging quality and imaging speed, which will facilitate the wide application of our SI-FLFM in biomedical research.
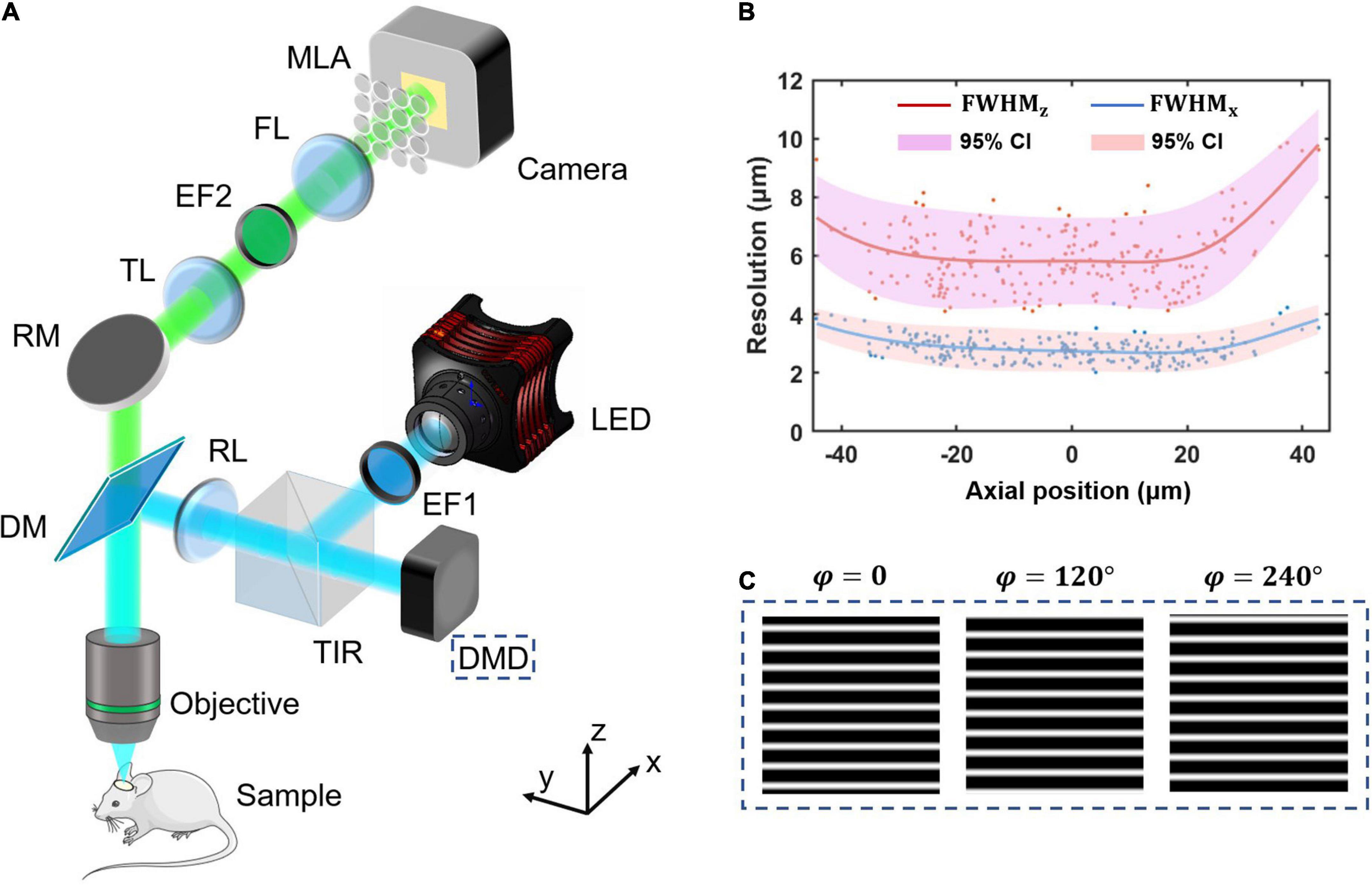
Materials and methods Optical designStructured-illumination Fourier light field microscopy is built by introducing structured illumination in the conventional FLFM. We firstly design a FLFM for biological imaging in vivo, and then introduce a digital micromirror device (DMD) into its illumination path to project the structured patterns. The key of designing FLFM is to assign spatial spectrum information to different microlens, resulting in multi-view imaging. And the three-dimensional (3D) reconstruction is carried out through the Richardson-Lucy deconvolution algorithm. Figure 1 shows the system design (same as that in our former report (Zhai et al., 2022)) and resolution calibration of the SI-FLFM.
FIGURE 1
Figure 1. The system design and resolution calibration of SI-FLFM. (A) Optical scheme of SI-FLFM. The digital micromirror device (DMD) plane is conjugated with the native objective plane (NOP), thus projects the structured patterns on the sample. EF1, excitation filter 1; DMD, digital micromirror device; TIR, total internal reflection prism; RL, relay lens; DM, dichroic mirror; RM, reflector mirror; TL, tube lens; EF2, emission filter 2; FL, Fourier lens; MLA: microlens array. (B) Three-dimensional resolution with 95% confidence interval (CI), fitted by LOESS algorithm. The resolution is calibrated by full-width-at-half-maximum (FWHM) of the imaging results of sub-resolution micro-beads (Φ = 1.1 μm). (C) The generated sinusoidal patterns of different phases for illumination are loaded in the memory of DMD. The fringe period is 90 pixels, calculated based on the DOF of SI-FLFM.
In the illumination path, we choose a collimated LED (SOLIS-470C, Thorlabs) for high-power excitation. The excitation light is filtered by the EF1 (Excitation Bandpass Filter, MF469-35, Thorlabs) to narrow the spectrum with a center wavelength at λex≈470nm, followed by being reflected to the DMD (1920 × 1080 pixels, DLP9500, TI) by a total internal reflection prism (TIR). The DMD consists of a micro-mirror array, in which each mirror can be mechanically rotated to achieve binarized projection. A dichroic mirror (DM, DMLP490, Thorlabs) is used to separate excitation and emission light. We then use two 4f relay systems, the first consisting of two doublets lens (f = 150 mm, AC508-150A and f = 300 mm, AC508-300A, Thorlabs, not shown in Figure 1A) and the second consisting of an RL (Relay Lens, f = 200 mm, AC508-200A, Thorlabs) and an objective (f = 7.2 mm, numerical aperture is 1.05, XLPLN25XWMP2, OLYMPUS). Thus, the surface of the DMD is conjugated to the native object plane (NOP) of the objective. When we load structured patterns of different phases (as shown in Figure 1C) on the DMD, they will modulate the in-focus sample in the object space correspondingly. The illumination path is designed as Köhler Illumination to perform uniform illumination.
In the detection path, the emission light from the sample is collected by the objective and then imaged by a tube lens (TL, f = 200 mm, AC508-200A, Thorlabs) at 27.78 times magnification. On the back focal plane of TL, we place an emission bandpass filter (EF2, customized, λem≈525nm, Edmund) to filter out the residual excitation light. We then use a Fourier lens (FL, f = 300 mm, AC508-300A, Thorlabs) to perform optical Fourier transform. On the back focal plane of FL, a microlens array (MLA, FEL-46S03-38.24PM, d = 3 mm, f = 38.24 mm, Sigma) is placed. Different microlens intercepts spatial spectra differently, performing the multi-view imaging on the camera (S-25A80 CoaXPress, Adimec).
Based on the optical design above, our system works at 3.54 times magnification and 0.139 NA for all 31 views (as shown in Figure 2) imaging. The maximum field of views (FOV) is up to Φ = 840 μm, the depth-of-field (DOF) is about 90 μm. The size of the surface of the DMD after being projected by the illumination light path is 1,493 μm × 840 μm, which can cover the entire FOV.
FIGURE 2
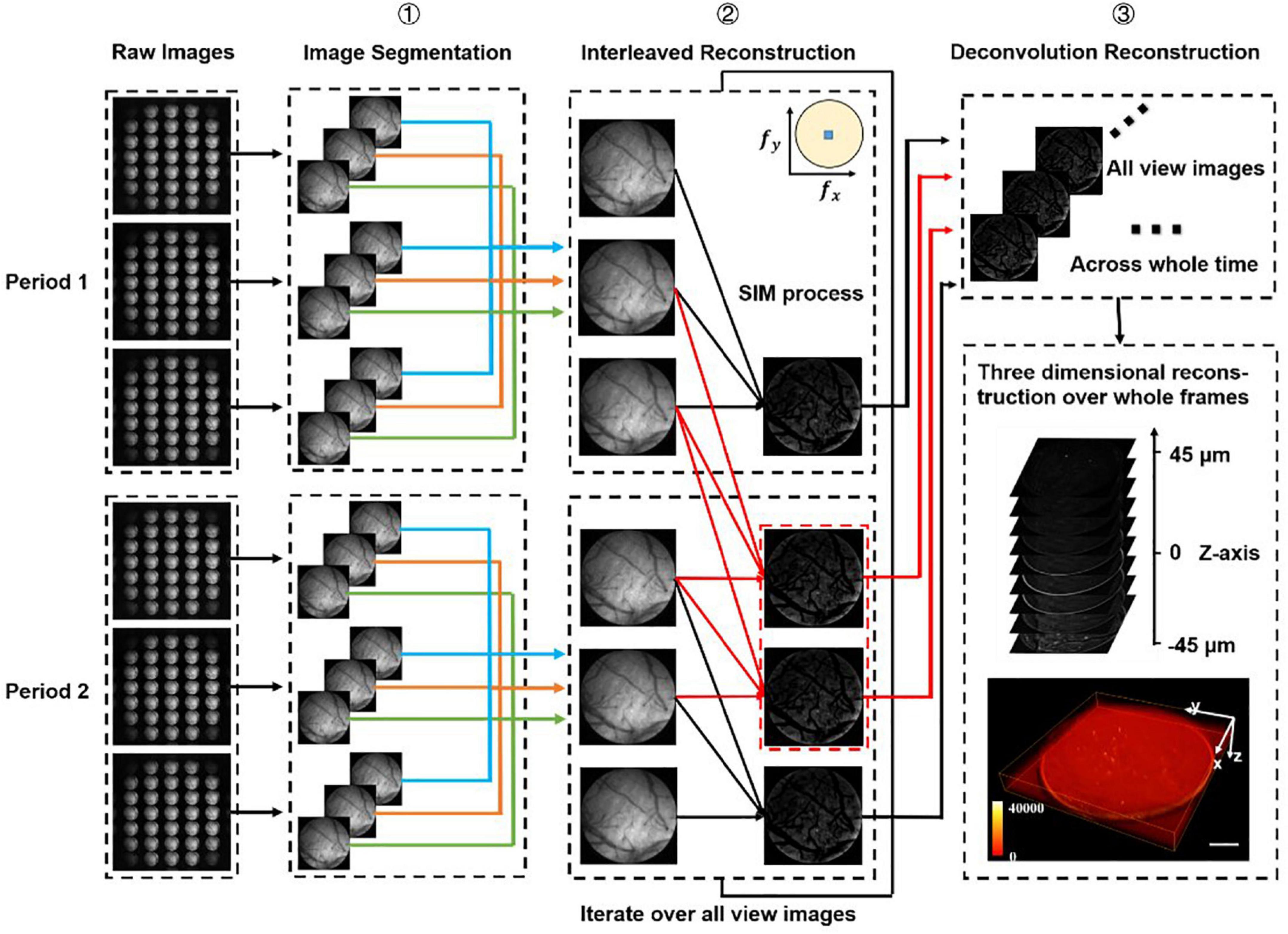
Figure 2. Data processing procedure. We firstly divide all the raw images (number: 3n) into different periods (number: n), and we have chosen two periods as an example to demonstrate the processing procedure. Step 1: Segment all raw images into sub-images at multi-views. Step 2: Use optical-sectioning algorithms and interleaved reconstruction to achieve optical-sectioning (OS) images without losing frames. The red lines indicate the adding frames recovered by interleaved reconstruction. The number of OS images is 3n-2 rather than n for each view. This step will iterate until all views sub-images have been processed. Step 3: 3D reconstruction for whole frames by the Richardson-Lucy deconvolution algorithm.
System calibrationTo demonstrate that our system can work at cellular resolution, we experimentally calibrate the resolution by imaging fluorescent beads (Φ = 1.1 μm, Thermo Fisher Scientific).
Due to the inevitable aberration in the system, it is difficult to achieve accurate reconstruction with the simulated point-spread-function (PSF) (as shown in Supplementary Figure 1a). Thus, we firstly calibrate the real PSF of the system by imaging the sparse fluorescence beads. To prepare the fluorescent beads sample for calibration, we dilute the original fluorescent beads solution by 2.5 × 105 times in agar at 95°C, then take a little of the solution on the glass slide and cool it to room temperature. The sample is sparse enough that only about 1 bead can be found in the whole FOV. After placing the sample, we move the stage (MT3/M-Z8, Thorlabs) at 1.5 μm steps and record the imaging results over a 90 μm range centered on the NOP. Based on these raw images, we obtain the PSFs of all views across the whole depth range (as shown in Supplementary Figure 1b) through the maximum connected domain extraction algorithm in MATLAB.
To calibrate the 3D resolution of the system, we then configure a 1:1000 dilution of fluorescent beads (Φ = 1.1 μm, Thermo Fisher Scientific) as sample. Through a single exposure of the sample, we obtain the raw image of a large number of fluorescent beads distributed in 3D space. Based on the previously acquired center coordinates of the sub-images for each view, we segment the original image to 31 individual images of different views. Then we use the extracted real PSFs and Richardson-Lucy algorithm to reconstruct the 3D image. We use the FWHM (full-width-at-half-maximum-intensity) to represent the resolution. However, off-axis aberrations (such as coma) distributed elsewhere in the FOV still can’t be avoided even with the real PSFs. Therefore, different locations in the FOV may show different resolutions. We use the Local Weighted Linear Regression (LOESS) algorithm (Hua et al., 2021) to fit the resolutions. Based on the linear least squares and first-degree polynomial model, the LOESS algorithm can be addressed by solving the optimization problem:
min(α(x0),β(x0))F(α(x0),β(x0))(1)
F(α(x0),β(x0))=∑i=1NKd(x0,xi)[yi-α(x0)-β(x0)xi]2(2)
Where Kd(x0,xi) is the weighting factor determined by the distance from xi to x0.
Kd(x0,xi)=exp(-(x0-xi)22d2)(3)
Here, we set the d to be 10 μm. The predicted resolution at x0 is y0=α^(x0)+β^(x0)x0. Based on the normal distribution, two standard deviations of the fit represent the 95% confidence interval. So, we calculate the standard deviation as:
σx0=F(α(x0),β(x0))∑i=1NKd(x0,xi)(4)
Thus, the 95% confidence interval at x0 is from y0−2σx0 to y0 + 2σx0. As shown in Figure 1B, the lateral resolution varies in 2∼4 μm as well as the axial resolution varies in 4∼10 μm in a 90 μm range DOF. The results suggest that our system works at cellular resolution.
System synchronizationStructured-illumination Fourier light field microscopy needs accurate system synchronization to perform structured illumination, which is achieved with a microcontroller (UNO Rev3, Arduino). As shown in Supplementary Figure 2, the microcontroller generates the digital signals, which are transmitted to the camera and DMD simultaneously. The camera starts an exposure once receiving a high-level signal (“1”), which will last for the same duration as that of the high-level signal. Meanwhile, the DMD will refresh the pattern to the next one after receiving a high-level signal. The refresh is so fast (about 20,000 Hz) that the settle time can be ignored. After the exposure, the camera needs about 12 ms to read out the image to RAM. Thus, the microcontroller is settled to a low level (“0”) signal for 12 ms to finish the transmission. In this way, we achieve a raw image under structured illumination.
Principles of structured-illumination Fourier light field microscopy and data processing proceduresIn SI-FLFM, we need to integrate the optical sectioning algorithm and the interleaved reconstruction algorithm into the Richardson-Lucy deconvolution. Here, we demonstrate the data processing procedure of two structure illumination periods in Figure 2.
Optical sectioning algorithmWe use I1,I2, and I3 to represent the images at different phases in a structured illumination period. Based on the modulating properties, the I1,I2 and I3 can be expressed by:
,,,]},,,]},,,]},,,,,]}],"socialLinks":[,"type":"Link","color":"Grey","icon":"Facebook","size":"Medium","hiddenText":true},,"type":"Link","color":"Grey","icon":"Twitter","size":"Medium","hiddenText":true},,"type":"Link","color":"Grey","icon":"LinkedIn","size":"Medium","hiddenText":true},,"type":"Link","color":"Grey","icon":"Instagram","size":"Medium","hiddenText":true}],"copyright":"Frontiers Media S.A. All rights reserved","termsAndConditionsUrl":"https://www.frontiersin.org/legal/terms-and-conditions","privacyPolicyUrl":"https://www.frontiersin.org/legal/privacy-policy"}'>
留言 (0)