
記住我










Diabetic retinopathy and diabetic macular oedema (DME) are the most common causes of preventable blindness worldwide,1–3 and the worldwide prevalence of DME is 4.7%–10.1%.4–6 Retinal thickening and hard exudates that threaten or involve the centre of the macula are considered to have clinical significance.7 8
For decades, focal and grid laser photocoagulation has been the mainstay treatment to prevent vision loss. However, the intraocular administration of antivascular endothelial growth factor (VEGF) agents reduces DME without tissue damage and is more effective in improving vision.1–3 Due to its advantages, currently, anti-VEGF agents has become the gold standard for the treatment of DME.
VEGF has been shown to have a major role in promoting exudation and neovascularisation,9–11 and the inhibition of VEGF can stabilise or even reverse the retinal damage due to DME.12 Over the past two decades, anti-VEGF agents such as pegaptanib, bevacizumab, ranibizumab and aflibercept were approved for the treatment of ocular diseases that involve retinal neovascularisation and exudation, including DME.13–17
Conbercept (KH902; Chengdu Kanghong Biotech Co., China) is a recombinant fusion protein with key domains 2, 3 and 4 from VEGF receptors 1 and 2. It has a high affinity for all VEGF isoforms and for placental growth factor.18 In 2013, it was approved in China for the treatment of neovascular (wet) age-related macular degeneration.18 In addition, conbercept is approved for the treatment of choroidal neovascularisation secondary to pathological myopia. The objective of the Sailing Study was to compare the efficacy and safety of intravitreal conbercept injections versus laser for the treatment of DME. This report includes the 1-year results of the Sailing Study and the 1-year results of its extension study in which patients crossed over to conbercept therapy.
MethodsStudy design and patientsThe Sailing Study was a multicentre, randomised, double-masked, double-sham, parallel controlled, phase III trial (registered at ClinicalTrials.gov) conducted at 18 centres in China. An open-label extension study was conducted after patients completed the Sailing Study.
For each patient, only one eye was enrolled in the study. If both eyes of a subject met the inclusion criteria, the investigators determined the target eye from the medical perspective, with the eye with poorer vision selected in principle. The key inclusion criteria were (1) >18 years of age; (2) type I or II diabetes mellitus; (3) haemoglobin A1c (HbA1c) of <10%; and (4) the study eye had to meet the following criteria: (i) DME involving the central fovea, (ii) ETDRS best-corrected visual acuity (BCVA) between 73 and 24 letters (Snellen equivalent of 20/40–20/320), (iii) central retinal thickness (CRT) of >300 µm according to optical coherence tomography (OCT) imaging, (iv) clear ocular media and adequate pupil dilation for examination and imaging and (5) the ETDRS BCVA of the subject’s non-target eye of ≥24 letters (equivalent to 20/320 of the Snellen vision). The key exclusion criteria were (1) active eye infection in either eye; (2) any ophthalmic conditions leading to macular oedema or alterations in vision other than diabetic retinopathy; (3) panretinal photocoagulation within 6 months prior to screening or local/grid retinal photocoagulation within 3 months prior to screening; (4) treatment with anti-VEGF drugs (eg, aflibercept, pegaptanib sodium, ranibizumab, bevacizumab, etc) within 6 months prior to screening; (5) any type of intraocular surgery (eg, cataract surgery, yttrium aluminium garnet (YAG) posterior capsulotomy, etc) within 3 months prior to screening; (6) uncontrolled hypertension; and (7) stroke, transient ischaemic attack, myocardial infarction or acute congestive heart failure within 6 months prior to screening. The detailed exclusion criteria are shown in the online supplemental appendix.
Randomisation and maskingDuring the Sailing Study, the eligible patients were randomly assigned in a 1:1 ratio to receive either sham laser followed by conbercept (sham/conbercept group) or laser followed by a sham injection (laser/sham group) according to the interactive web response system. Masking was performed for the patients, masked investigators and statisticians. Treatments were performed by unmasked investigators who were not involved in any other study work. The extension study was an open-label study, so no masking was necessary.
TreatmentsDuring the Sailing Study, the patients in the sham/conbercept group received sham laser and then an intravitreal injection of conbercept (0.5 mg, Chengdu Kanghong Biotech Co.) on day 0, followed by pro re nata (PRN) conbercept treatments and sham laser treatments during the monthly follow-up per predefined criteria. Patients in the laser/sham group received modified ETDRS grid photocoagulation and then a sham intravitreal injection on day 0. Starting at month 3, the laser group received PRN sham injections and active laser treatments during the monthly follow-up per predefined criteria, but the retreatment of laser will not be evaluated if less than 12 weeks from the last sham or active laser treatment. When the patients received active PRN conbercept or laser, the sham laser or sham injection was performed on the same day. An interval of at least 2 hours was ensured between the active or sham laser treatment and the active or sham intravitreal injection.
PRN laser was performed if any of the following criteria was met: (1) retinal thickening within a radius of 500 µm around the centre of the macula; (2) hard exudates with adjacent retinal thickening within a radius of 500 µm around the centre of the macula (for hard exudates left behind after retinal thickening regression, no treatment is required); (3) the total area of one or multiple regions with retinal thickening is no less than that of one optic disk (2.54 mm2); and the distance from a part of the region of retinal thickening to the centre of the macula is below a diameter of the optic disk (1800 µm).
The criteria for repeated conbercept treatments were (any of the following criteria satisfied): (1) CRT increased by at least 50 µm if compared with the previous minimum value; (2) CRT of ≥300 µm; (3) cystoid degeneration of retina, subretinal fluid or pigment epithelial detachment in the macular region; (4) ETDRS BCVA was improved by at least five letters if compared with that during the previous visit; and (5) ETDRS BCVA declined by at least five letters if compared with the maximum number of letters previously, combining with CRT increase, in comparison with that when ETDRS BCVA is at its optimum level.
Other treatments forbidden from the study included (1) anti-VEGF drugs other than conbercept; (2) any other antiangiogenic drugs; (3) ocular or systemic steroids; (4) drugs with toxicity to the lens, retina or nerves; (5) anticoagulant or antiplatelet therapy, except for low-dose prophylactic use; or (6) treatments to the study eye with impact on the efficacy and safety assessments of the present study, such as panretinal photocoagulation, verteporfin photodynamic therapy, external beam radiotherapy, vitrectomy, transpupillary thermotherapy and macular surgery.
Starting at month 6, whether the patients needed rescue treatment was assessed monthly. If patients met either one of the criteria for rescue treatment: (1) ETDRS BCVA declined by at least 15 letters compared with the maximum number of letters previously and lowered to a degree below the baseline; or (2) ETDRS BCVA decreased to a level that at least 10 letters fewer than the baseline during visits of two consecutive months, and the investigators believed that the visual impairment was caused by the persistent or worsening oedema from the DME, then their sham designation was switched to active treatment in whatever group they were assigned.
During the extension study, patients in both groups received PRN conbercept treatment with monthly follow-up.
AssessmentThe primary endpoint of the Sailing Study was the mean change in BCVA from baseline to month 12. Secondary endpoints included the change in CRT from baseline to month 12 and safety. Safety assessments included both ocular and non-ocular adverse events (AEs) and serious adverse events (SAEs). Other endpoints included changes in CRT, total macular volume (TMV), fluorescein angiographic leakage area, BCVA and the total score of 25-Item National Eye Institute Visual Function Questionnaire (NEI VFQ-25) from baseline to month 12, as well as the proportion of ≤5, 10 and 15 letters vision gain or loss from baseline to months 6 and 12.
The primary outcome of the extension study was the mean change in BCVA from month 12 to 24. Secondary outcomes included the long-term safety of conbercept, change in BCVA from month 12 to 24, change in CRT from month 12 to 24, change in TMV from month 12 to 24, change in leakage area from month 12 to 24 and the number of injections in the extension study.
Patients were followed up monthly for ophthalmological examinations, including ETDRS protocol BCVA, intraocular pressure, slit-lamp examination and OCT. Colour fundus photography (CFP) and fundus fluorescein angiography (FFA) were performed every 3 months. NEI VFQ-25 was completed every 6 months. Protocol BCVA was measured by using the standard ETDRS visual acuity chart (starting at the 4 m testing distance). All study-related image aqcuisitions were certificated by EyeKor, LLC (online supplemental table 1). OCT, CFP and FFA images were assessed by the reading centre of the University of Wisconsin–Madison.
Statistical analysisAssuming that the changes in BCVA from baseline to month 12 had a difference of five letters between the two groups, the SD of the two groups was 10 and 12 letters, respectively. A total of 208 patients (104 in each group) were calculated with a one-sided significance level of 2.5% and a power of 90%. Assuming the drop-out rate of 20%, 124 patients were needed in each group.
The full analysis set in the Sailing Study included all randomised patients who received at least one treatment with the corresponding efficacy assessment. The safety set (SS) in the Sailing Study included all patients who received at least one treatment and at least one safety assessment. The intention-to-treat population in the extension study included all patients who agreed to continue the follow-up and received at least one extension assessment. The SS in the extension study included all patients who received at least one conbercept treatment during the 2-year period.For the primary endpoint of the Sailing Study, the missing values were imputed using the last observation carried forward (LOCF) method. For patients who had a rescue treatment, the missing values were imputed using LOCF method prior to the first rescue treatment. Missing values for the secondary endpoints in the Sailing Study and all outcomes in the extension study were not imputed.
Statistical analyses were performed using SAS V.9.4. Continuous variables were presented as means±SD. The analysis of covariance was used for the primary outcome comparison. For secondary efficacy outcomes, the independent t-test was used for intergroup comparisons and a paired t-test was used for comparisons between two time points. Categorical variables were presented as frequencies (percentages) and compared with the Cochran-Mantel-Haenszel χ2 test or Fisher’s exact test, as appropriate. A two-sided p value of <0.05 was considered statistically significant.
ResultsPatient disposition, baseline characteristics and treatment experienceBetween August 2014 and December 2015, a total of 251 eligible patients were randomised. After completing the Sailing Study, 157 patients enrolled in the extension study. Figure 1 shows the study flowchart and analysis sets.
 Figure 1
Figure 1 Study flowchart.
The baseline characteristics of the patients in the Sailing Study were well balanced between groups (table 1). The patient disposition in the extension study is shown in online supplemental table 2. Eyes in the Sailing Study received an average of 2.6 and 2.7 sham or laser treatments in the sham/conbercept group and laser/sham groups, respectively, and 9.5 and 9.7 intravitreal or sham injections, respectively. More patients in the laser group required a rescue treatment compared with the conbercept group in the Sailing Study (20.3% vs 4.0%, p<0.001). During the extension study, the numbers of conbercept treatments were 8.5±3.5 and 8.6±3.4 in the conbercept and laser groups, respectively (online supplemental table 3).
Table 1Baseline characteristics of patients in the Sailing Study
EfficacySignificant improvement in BCVA from baseline to month 12 was observed in the conbercept group (8.2±9.5 letters, p<0.001), whereas no improvement was observed in the laser group (0.3±12.0 letters, p=0.810) (figure 2A). The changes in BCVA from baseline were significantly different between the two groups at all time points during the first year (all p<0.001). The subset of eyes that continued in the extension study also showed a similar result during the first year (figure 2B). During the extension study, after patients in the laser group crossed over to receiving PRN conbercept treatment, there was a significant improvement in BCVA at all time points of the second year (vs month 12, all p<0.05) (figure 2B). The change in BCVA at month 24 in the laser group was 4.9±9.4 letters from month 12 (p<0.001) and 8.0±11.4 letters from baseline (p<0.001). For patients in the conbercept group, 2-year results showed that they continued to maintain visual acuity gains from intravitreal injection.The changes in BCVA at month 24 in the sham conbercept group were 2.3±8.8 letters from month 12 (p=0.030) and 8.3±12.4 letters from baseline (p<0.001).
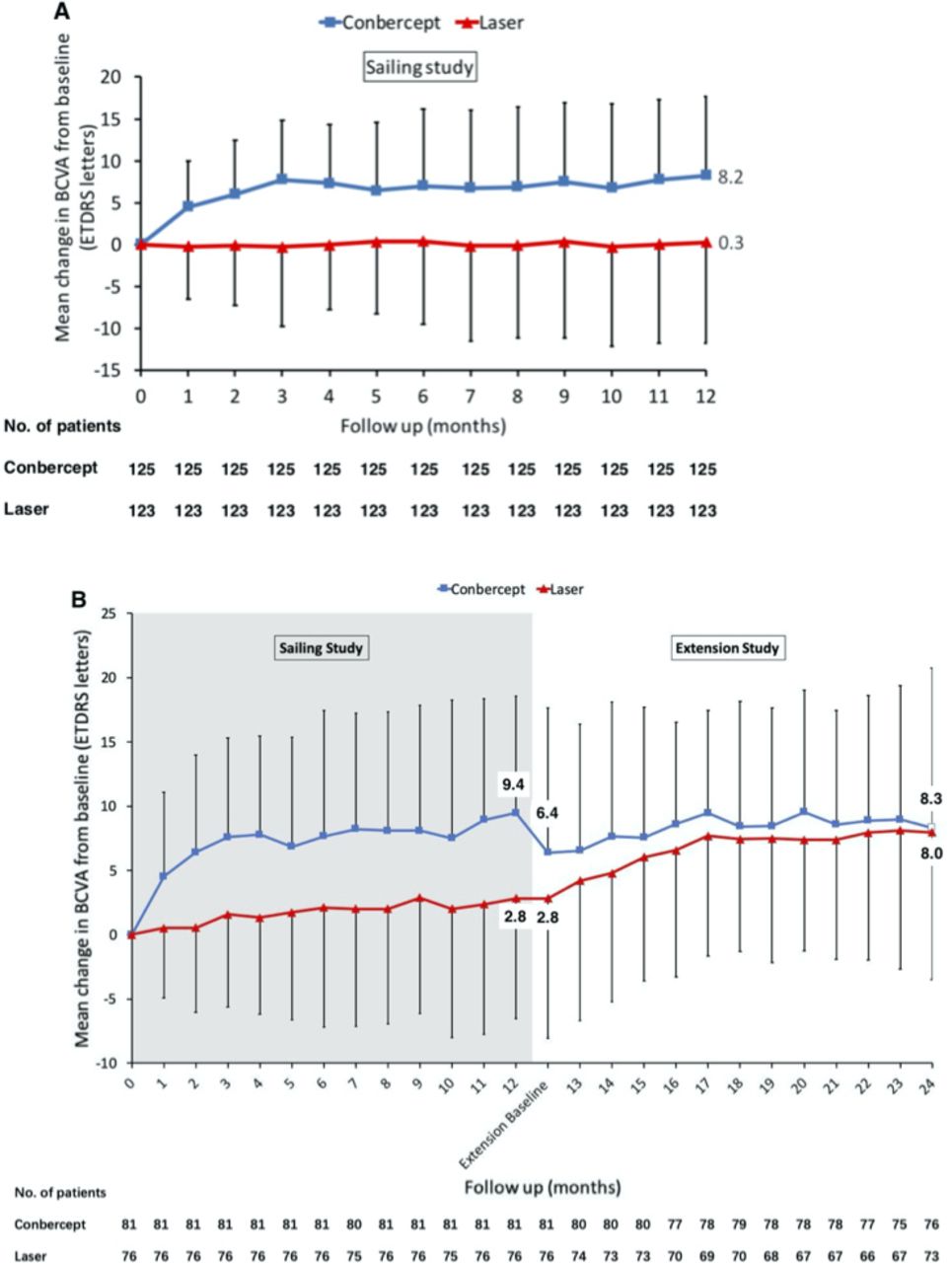 Figure 2
Figure 2 Mean changes in BCVA for laser and conbercept study groups in the Sailing Study. (A) One-year results compared with baseline. (B) Two-year results compared with baseline for the subset of eyes that continued in the extension study. Error bars denote SD. BCVA, best-corrected visual acuity.
Both the results of 6 months and those of 12 months showed that significantly more eyes treated with conbercept gained vision from baseline, whereas significantly more eyes treated with laser lost letters from baseline (all p<0.001) (online supplemental table 4). The proportion of eyes with vision gain of ≥15 letters from baseline were 17.4% (21/121) and 25.0% (28/112) at months 6 and 12 in the sham/conbercept group compared with 6.8% (8/118) and 14.9% (13/87) in the laser/sham group, respectively, at the same time points. The proportion of eyes with vision loss of ≥15 letters from baseline were 1.7% (2/121) and 3.4% (4/112) at months 6 and 12 in the sham/conbercept group compared with 0% (0/118) and 1.1% (1/87) in the laser/sham group, respectively.
Significant differences in CRT were observed beteween the two groups at all time points in the first 12 months (all p<0.01) (figure 3A). The mean reduction of CRT from baseline to month 12 was 200±210 µm (p<0.001) in the sham/conbercept group and 130±190 µm (p<0.001)in the laser/sham group. Significant reduction of TMV from baseline was observed by month 3 (1.3±0.9 mm3, p<0.001) in eyes treated with sham/conbercept, whereas this reduction was observed at month 6 (0.6±0.7 mm3, p=0.004) in eyes treated with laser/sham (figure 3C). There was a significant difference in TMV between the two groups at months 3 and 6 (both p<0.05), but not at months 9 and 12. The trends of the leakage area changes were similar to those of the CRT changes. The mean reduction of leakage area from baseline to month 12 was 7.9 mm2 (p<0.001) in the sham/conbercept group and 3.9 mm2 (p<0.001) in the laser/sham group, and the leakage area was significantly decreased in the sham/conbercept group compared with the laser/sham group (p<0.001). The mean change in the NEI VFQ-25 total score from baseline was significantly different between the sham/conbercept group and the laser/sham groups at months 6 (4.4±16.0 vs −1.5±15.5, p=0.004) and 12 (4.3±19.5 vs −3.8±17.7, p=0.001) (figure 3E). During the extension study, the improvements in anatomical outcomes (CRT, TMV and leakage area) were maintained in both groups (figure 3B,D), and the laser group did better in reducing fluorescein leakage area.
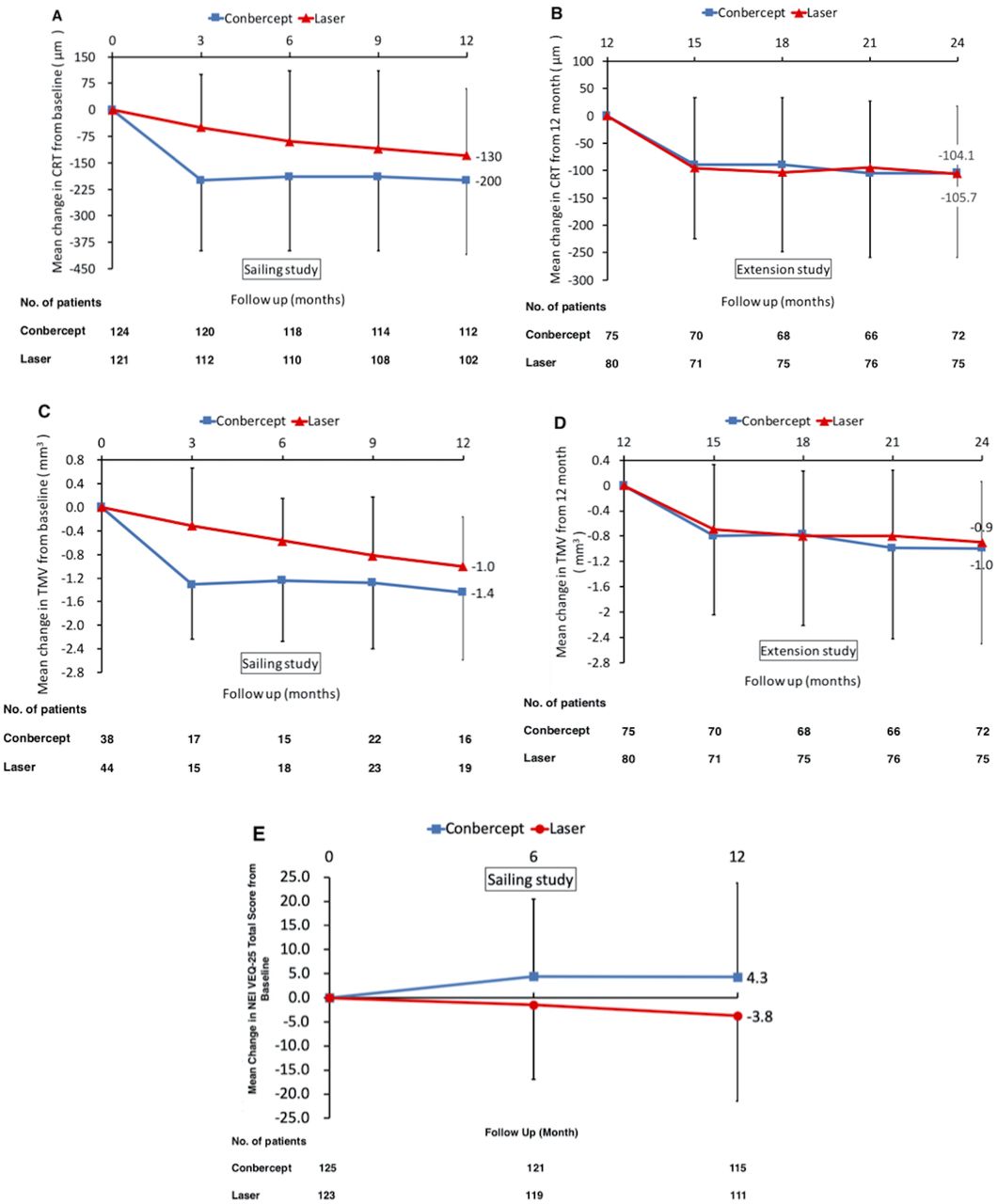 Figure 3
Figure 3 Mean changes of secondary endpoints for laser and conbercept study groups in the Sailing Study. (A) One-year results of mean CRT changes compared with baseline. (B) Extension study results of mean CRT changes from 12 to 24 months. (C) Mean changes in TMV compared with baseline (1-year results). (D) Mean changes in TMV from 12 to 24 months (extension study results). (E) Mean change in NEI VFQ-25 total score from baseline to month 12 of the Sailing Study. Error bars denote SD. CRT, central retinal thickness; NEI VEQ-25, 25-Item National Eye Institute Visual Function Questionnaire; TMV, total macular volume.
SafetyIn the Sailing Study, the proportion of total AEs (87.2% vs 87.1%) and SAEs (17.6% vs 19.4%) were similar between the sham/conbercept and laser/sham groups (table 2). The most common AEs were visual impairment (20.0%), upper respiratory tract infection (20.0%), intraocular hypertension (12.8%), hypertension (12.0%), elevated blood pressure (12.0%) and subconjunctival haemorrhage (12.0%) in patients treated with conbercept. The most common AEs were visual impairment (19.4%), upper respiratory tract infection (17.7%), hypertension (10.5%) and diabetic nephropathy (10.5%) in patients treated with laser. More patients reported ocular AEs (57.6%) in the sham/conbercept group compared with 39.5% in the laser/sham group. Most of the additional ocular AEs with conbercept treatment were intraocular hypertension (12.8% vs 4.8% with laser) and subconjunctival haemorrhage (12.0% vs 3.2% with laser). Endophthalmitis occurred in two patients treated with conbercept and one thereof was non-infectious intraocular inflammation. Endophthalmitis did not occur in patients treated with laser. The incidence of non-ocular SAEs in the laser/sham group (16.9%) was higher than in the sham/conbercept (12.0%). The incidences of Anti-Platelet Trialists’ Collaboration-defined arterial thromboembolic events (APTC-ATEs) were equal (3.2%) in both groups.
Table 2AEs in the Sailing Study and extension study
The AEs that occurred during the extension study were similar to those in the Sailing Study. Intraocular hypertension (24/156, 15.4%) and upper respiratory tract infection (24/156, 15.4%) were the most frequently reported AEs in the second year. For APTC-ATEs, rates were 7.7% (12/156) in the extention study. No death occurred in the conbercept group during the 2-year period. One patient in the laser group died from cerebral haemorrhage during the first year, and another patient died from ischaemic cardiomyopathy in the second year.
DiscussionThe Sailing Study was designed to evaluate the use of conbercept for the management of patients with centre-involved DME. The primary endpoint of the Sailing Study was the mean change in BCVA from baseline to month 12. The primary outcomes of the extension study were safety and mean change in BCVA from month 12 of the Sailing Study to month 12 of the extension study. The results of the study show that conbercept can improve the BCVA in patients with centre-invovled DME, and its efficacy was better than that of traditional ETDRS laser photocoagulation. The safety of conbercept was good with the total occurrence of AEs being similar between the two groups.
In the present study, the sham/conbercept group followed a PRN regimen of conbercept injections after the first injection at baseline and over 12 months, the actual number of injections was 9.5 with a mean improvement of +8.2 letters. The results of the 1-year open-label extension study showed that the efficacy of conbercept was maintained for 24 months, but the number of injections was not significantly reduced. In the extension study after 12 months, patients in the laser/sham group were crossed over and received conbercept PRN. The laser/sham group showed a significant improvement in BCVA after crossing over to conbercept therapy, and there was no difference in BCVA between the two groups at the end of the extension study. This is a different result from other studies where anti-VEGF was delayed and the delayed anti-VEGF group did not catch up to the group treated with anti-VEGF from baseline.
Several studies have demonstrated that anti-VEGF treatment is superior to laser in DME.15–17 19 The RISE and RIDE trials compared the efficacy of ranibizumab for DME with PRN laser photocoagulation, and the results showed that ranibizumab improved BCVA, reduced the risk of vision loss and improved the parameters of macular oedema.19 The VISTA and VIVID studies examined two regimens of aflibercept versus laser photocoagulation for DME and showed that aflibercept was superior to laser photocoagulation in terms of visual acuity and anatomic changes.15–17 Despite using a different anti-VEGF agent, the Sailing Study showed similar results to the RISE, RIDE, VISTA and VIVID trials. Conbercept, which is similar to aflibercept, is a recombinant fusion protein composed of VEGF binding domain from human VEGF receptors 1 and 2.20 However, conbercept may have a higher potency and a longer half-life compared with aflibercept because of an additional portion in the fourth binding domain of VEGF receptor 2, which enhances the rate of binding and the stability of the binding complex.21 In the 12-month results of aflibercept in the phase III studies, the mean injection numbers of 2q4 and 2q8 groups were 11.8 and 8.4 in VISTA, and 12.2 and 8.7 in VIVID, respectively.15 In our Sailing Study, the number of conbercept injections was 9.5, which is similar to the number of injections in 2q8 group. While this number is lower, it is important to recognise that the injection freuqnecy in VIVID and VISTA were fixed not PRN. Of note, at the time of the study, aflibercept and ranibizumab had not been approved for the treatment of DME in China. Thus, additional clinical studies are needed to compare the efficacy of conbercept with other anti-VEGF agents in DME.
The efficacy of laser photocoagulation in improving BCVA during the Sailing Study was similar to that in VISTA and VIVID, with +0.3±12.0,+0.2±12.5 and+1.2±10.6 letters in BCVA, respectively. In our study, the percentage of eyes that had BCVA improvement in the laser group was greater than similar laser groups in other clinical trials. The reason for this is unknown but may depend on external factors such as the investigators’ operational approaches and the type of laser used, as well as the patient population. The factors associated with differences in visual acuity outcomes in eyes treated with panretinal photocoagulation include the HbA1c levels and the severity of the diabetic retinopathy in these patients.22 However, our results do indicate that the standard treatment of laser photocoagulation is still a good therapeutic option for some patients, but it is noteworthy that the laser group had a higher proportion of eyes with vision loss than the conbercept group. However, the proportion of patients with loss of ≥15 letters in the laser therapy group was 1.1%, far less than the 9.1% and 10.6% observed in the VIVID and VISTA trials.15–17
During the extension study, patients in the laser group had significant improvement in visual acuity after crossing over to receive PRN conbercept injections and reached similar mean change in BCVA levels compared with patients in the conbercept group at 24 months, suggesting the promising efficacy of conbercept in DME. However, it took 2 months to demonstrate a four-letter BCVA improvement in the laser group, while the conbercept group achieved it in the first month. The delayed benefit from conbercept injection in the laser group could be due to the longer duration of the oedema and resulting outer retinal changes due to the prolonged oedema in the macular tissue. Nonetheless, compared with the RESTORE extension study of ranibizumab where it took nearly 12 months to gain +4 letters after switching to ranibizumab,23 conbercept cross-over patients exhibited a faster visual acuity improvement. The rapid and sustained effect of conbercept could improve the quality of life and reduce the risk of vision loss for patients with DME.
With respect to the anatomical outcomes (CRT, TMV and leakage area) and the vision-related quality of life scores (NEI VFQ-25 total score), rapid and sustained improvements were observed in the conbercept group over the first 12 months. The laser group showed worse vision-related quality of life results, despite steady but slow improvement in anatomical indices. In the extension study, CRT in the laser group decreased to a similar level as that in the conbercept group. This supports the fact that conbercept improved the pathological changes in the eye, resulting in better visual acuity and better vision-related quality of life. This was also observed in the RISE, RIDE, VISTA, VIVID and RESTORE trials.15–17 19 23
In the Sailing Study, the major AEs in the conbercept group were intraocular hypertension and subconjunctival haemorrhage (mostly mild), which were consistent with the AEs reported in previous studies.15–20 23–25 Endophthalmitis occurred in two patients after the injection of conbercept, which was aligned with the low incidence of endophthalmitis reported after the injection of saline,26 bevacizumab,27 ranibizumab19 28 and aflibercept.17 The incidence of non-infectious intraocular inflammation was 0.12% (1/861) of all injections, within the range of 0.09%–0.37% reported in the literature.29 These AEs were predominantly associated with any routine intravitreal injection. The 2-year results of safety were similar to the safety profile of conbercept in the 1-year Sailing Study. No new ocular and non-ocular AEs were identified. Overall, the safety of conbercept treatment was similar to other anti-VEGF agents.
A limitation of the extension study is that only 62.6% (157/251) of the initial patients entered the study. Of the 157 subjects enrolled, 142 completed the extension study and 15 withdrew before completing the study. Due to the limited sample size, it is hard to identify the significance of infrequent SAEs that occurred across groups, such as cerebrovascular accident and myocardial infarction.
In conclusion, the 0.5 mg conbercept PRN treatment regimen significantly improved the functional and anatomical outcomes of patients with centre-involved DME, compared with laser photocoagulation. The evidence from the Sailing extension study confirms that conbercept was well tolerated and its efficacy was sustained through 24 months. Intravitreal injections of conbercept can be considered an additional option in the management of patients with DME.
留言 (0)