
記住我










There are many cases of hypertrophy, and of great hypertrophy of the heart, in which during life and after death no source of increased work can be discovered. There is still much that remains to be explained. —Thomas Lewis (Diseases of the Heart, London: Macmillan and Co, 1933)
In the beginning…The last century has seen great advances in cardiology, including cardiac catheterisation, coronary bypass surgery, valve intervention, transplantation, electrophysiology and imaging. Advances in the field of heart muscle disease have paralleled these developments; this review focuses on hypertrophic cardiomyopathy (HCM) as an exemplar in light of the particular influence British cardiology has had on improvements in its diagnosis, symptom management and prevention of disease-related complications.
Erudite physicians and pathologists have described individual cases of thick and heavy hearts for centuries,1 but it was only in the middle of the 20th century that HCM emerged as a discrete clinical entity, with almost simultaneous reports of the characteristic clinical, pathological and physiological abnormalities that define the disease to this day. In the context of this anniversary edition, it is appropriate to highlight the now famous paper by pathologist Donald Teare (figure 1), published in the British Heart Journal in 1958, in which he reported eight cases of asymmetrical hypertrophy of the interventricular septum.2 While this was not, as is popularly believed, the first description of HCM, it was one of the first papers to correlate clinical and electrocardiographic abnormalities with a detailed macroscopic and microscopic examination of the heart, thereby establishing features such as myocyte disarray, mitral valve abnormalities and myocardial fibrosis as cardinal features of the disease. Remarkably, in an addendum, Teare2 reported a ninth case who had died while riding his bicycle and was a brother of one of the eight already described individuals; his surviving younger sister had also just attended the outpatient department of Hammersmith Hospital in London with signs compatible with left ventricular hypertrophy.
 Figure 1
Figure 1 Photograph of Donald Teare (1917–1979), a well-liked teacher, colleague and much respected forensic pathologist. By 1951, he had already performed more than 25 000 autopsies. Figure reproduced from Medico-legal Journal.57
At the time of Teare’s paper, many physicians were characterising another typical feature of the disease, dynamic left ventricular outflow tract obstruction. This was probably first described in Paris by Vulpian and colleagues3 in the 19th century and was, by virtue of its typical clinical manifestations, the focus of attention for many of the key players in the field for the next 60 years, including John Goodwin, Celia Oakley, Paul Wood, Eugene Braunwald, Douglas Wigle, Barry Maron, William McKenna and many others (figure 2).4 Russell Claude Brock5 of Guy’s Hospital in London is often credited as the first to recognise ‘functional obstruction of the left ventricle’ in vivo on the operating table, but it was William Cleland at the Hammersmith Hospital in London who performed the first septal myotomy in 1957. After visiting Cleland in London, it was Glenn Morrow–who incidentally also had the disease–who developed the surgical procedure that is still used today to relieve severe symptomatic left ventricular outflow tract obstruction.
 Figure 2
Figure 2 Timeline of notable contributions to the characterisation of HCM and its subsequent management. Due to space constraints, this list is far from exhaustive; the authors acknowledge that not all of the many brilliant researchers during this period have been referenced. *John Goodwin and Carolyn Biro subsequently founded the Hypertrophic Cardiomyopathy Association in 1990, emphasising the importance of providing a platform for guidance and counselling to help patients and their families better understand their disease and to cope with its psychological and emotional burden. Much of this work is now being undertaken by Cardiomyopathy UK. ACC, American College of Cardiology; AHA, American Heart Association; ESC, European Society of Cardiology; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter-defibrillator.
EpidemiologyAs noted by Wallace Brigden6 in 1957 when he used the term cardiomyopathy for the first time, ‘The true incidence of these diseases is unknown but is probably higher than is generally supposed….’ Numerous studies in healthy adults suggest a prevalence of around 1:500, with no differences between ethnicities or geographical locations. Extrapolation from genetic sequencing studies suggests that the frequency of potentially pathogenic sarcomeric protein gene variants may be even higher.7 As a recent study in this journal has shown, estimates from electronic health records indicate that the prevalence of clinically evident disease may be closer to 1:2500.8 A male predominance of around 60% is a constant finding, with women being older at presentation, more symptomatic and with a greater degree of left ventricular outflow tract obstruction. Importantly, women may have higher all-cause mortality probably related to more heart failure and stroke-related deaths.9
A familial diseaseIn 1949, William Evans,10 a London cardiologist, described five patients from three families with idiopathic left ventricular hypertrophy, and with Donald Teare’s paper and the report in 1961 of a French-Canadian family from Quebec in whom 30 members across five generations were found to have HCM,11 the genetic basis of HCM was established. It was not until 1989, however, that an exhaustive genetic linkage analysis resulted in the discovery of a locus on chromosome 1412 that was subsequently found to harbour a disease-causing variant in the gene encoding beta-myosin heavy chain (MYH7), one of the principal components of the cardiac sarcomere.13 14 Since then, many more causative variants in MYH7 and other sarcomeric and non-sarcomeric genes have been discovered, with MYH7 and MYBPC3 accounting for 60%–70% of individuals with a positive test.
With advances in rapid genetic sequencing, genotyping is now part of routine practice and not only helps to confirm the diagnosis but also assists in the diagnosis of important phenocopies such as transthyretin cardiac amyloidosis15 and Anderson-Fabry disease.16 This is pertinent because treatment strategies differ widely depending on the underlying disease substrate17 and risk models are not transferable.18
Genotyping has also transformed our ability to perform presymptomatic cascade screening of first-degree relatives, identifying at-risk individuals who warrant close surveillance. Recent data suggest that if relatives of an index patient carry the same pathogenic sarcomeric gene variant, around half will go on to develop HCM over 15 years of follow-up19; crucially, it is only when those individuals develop the phenotype that they are subject to the risk of developing disease-related complications.
Genetic testing can also provide information surrounding prognosis among specific HCM cohorts. For example, linkage analyses demonstrating mutations in cardiac troponin T have often been characterised by relatively mild hypertrophy but a high incidence of sudden death,20 while mutations involving the converter region of MYH7 have been associated with a higher risk of lethal arrhythmia and progression to heart failure.21 Similarly, a large cohort study published in this journal of consecutive HCM probands screened with high-throughput sequencing of 41 genes demonstrated a class effect for sarcomeric protein variants with a strong influence on clinical presentation, left ventricular morphology and survival.22
Management of left ventricular outflow tract obstructionThe existence of dynamic left ventricular outflow obstruction as a clinical entity was hotly debated in the early 1960s,23 24 but its importance was largely settled in the 1980s with the demonstration of genuine impedance of left ventricular ejection at the level of the outflow tract. From the outset, it was noted that many patients did not have evidence of outflow tract obstruction at rest but could develop it during physical exertion or with simple bedside manoeuvres such as the Valsalva.25 26
During the long history of HCM, many invasive and non-invasive treatment strategies to ameliorate or eliminate left ventricular outflow tract obstruction have been proposed. Negative inotropic agents (β blockers, calcium antagonists and disopyramide) remain first-line therapy, but while often effective in latent obstruction they tend to be less so in patients with severe resting obstruction. Some caution is advised with the use of calcium antagonists, particularly verapamil, as they can unpredictably increase the obstruction due to vasodilatation. The type 1A antiarrhythmic agent disopyramide can decrease or even abolish the obstruction but has the disadvantage of anticholinergic side effects and tachyphylaxis in a substantial proportion of patients.
In patients with drug-refractory symptoms, invasive reduction of the thickness of interventricular septum may be necessary. The original Morrow procedure or variations thereof, in which a small section is removed from the interventricular septum via an aortic approach, remains the reference standard, and in selected high-volume expert centres this procedure is safe and effective in more than 75% of patients.27 28 It is also associated with a low annualised mortality, although other disease-related complications such as atrial fibrillation and left ventricular systolic impairment may still occur in the long term.29
Alcohol septal ablation (ASA) was first introduced by Sigwart and Kühn in the 1990s as an alternative to surgical myectomy.30–32 The technique aims to produce a limited infarct in the upper interventricular septum through instillation of alcohol via selective cannulation of one or more of the septal perforator vessels. The procedure was initially associated with a high risk of complications including complete heart block, but refinement of the technique including routine intraprocedural myocardial contrast echocardiography to help limit infarct size and the inclusion of preprocedure CT coronary angiography to confirm an appropriate septal branch of the left anterior descending artery (figure 3), thereby minimising the number of aborted procedures, have improved safety and efficacy.33 Meta-analyses of long-term outcomes after septal reduction therapy showed a slightly higher LVOT gradient following ASA compared with myectomy, but with no significant differences between the two procedures in functional parameters.34 Importantly, as with surgery, complication rates relate to the case volume and expertise.35
 Figure 3
Figure 3 Example of CT angiography identifying the septal branch required to provide optimal infarct location. (A) Preprocedure transthoracic echocardiogram in end-systole showing the septal contact from the anterior mitral valve leaflet resulting in (B) a maximal LVOT gradient on Valsalva of 96 mm Hg. (C) Preprocedure CT angiogram. The traced septal vessels from two-dimensional images were projected onto the coronary angiogram ‘map’. This CT angiogram was rotated to minimise foreshortening and remove overlap (in this example to RAO cranial). (D) The equivalent invasive angiogram projection. The target artery is identified and only this sub-branch is occluded for alcohol delivery. (E) Transthoracic echocardiogram performed 4 months after alcohol septal ablation showing absence of septal contact in end-systole and (F) maximal provocable LVOT gradient of only 13 mm Hg. C and D were provided courtesy of Dr Rob Cooper, Liverpool Heart and Chest. LVOT, left ventricular outflow tract; RAO, right anterior oblique; RV, right ventricular.
In medically refractory symptomatic patients who are suboptimal for septal reduction therapy, DDD pacing with a short atrioventricular delay has been proposed as an alternative strategy and until recently was one of the few interventions in HCM subjected to a randomised clinical trial.36 These trials demonstrated reductions in LVOT gradient but reported conflicting results on improvement in quality of life and symptoms.37 38
Risk stratification and prevention of sudden deathIn 1929, CH Whittle,39 a consultant pathologist at Addenbrooke’s Hospital, reported on a previously healthy 20-year-old University of Cambridge student dying suddenly while bike riding. On autopsy he was found to have an ‘enormous heart’ with excessive hypertrophy, particularly of the left ventricle. Sudden cardiac death (SCD) is a tragic, potentially avoidable but often unpredictable complication of HCM that may present as the first manifestation of this disease (figure 4). Contemporary studies demonstrate that overall survival rates have improved progressively in the modern era, such that the annual SCD rate now ranges from as low as 0.3% to 1.0%40 41 in referral populations. However, population-based data provide powerful evidence that it remains a significant issue along with atrial fibrillation, heart failure and stroke.41 42
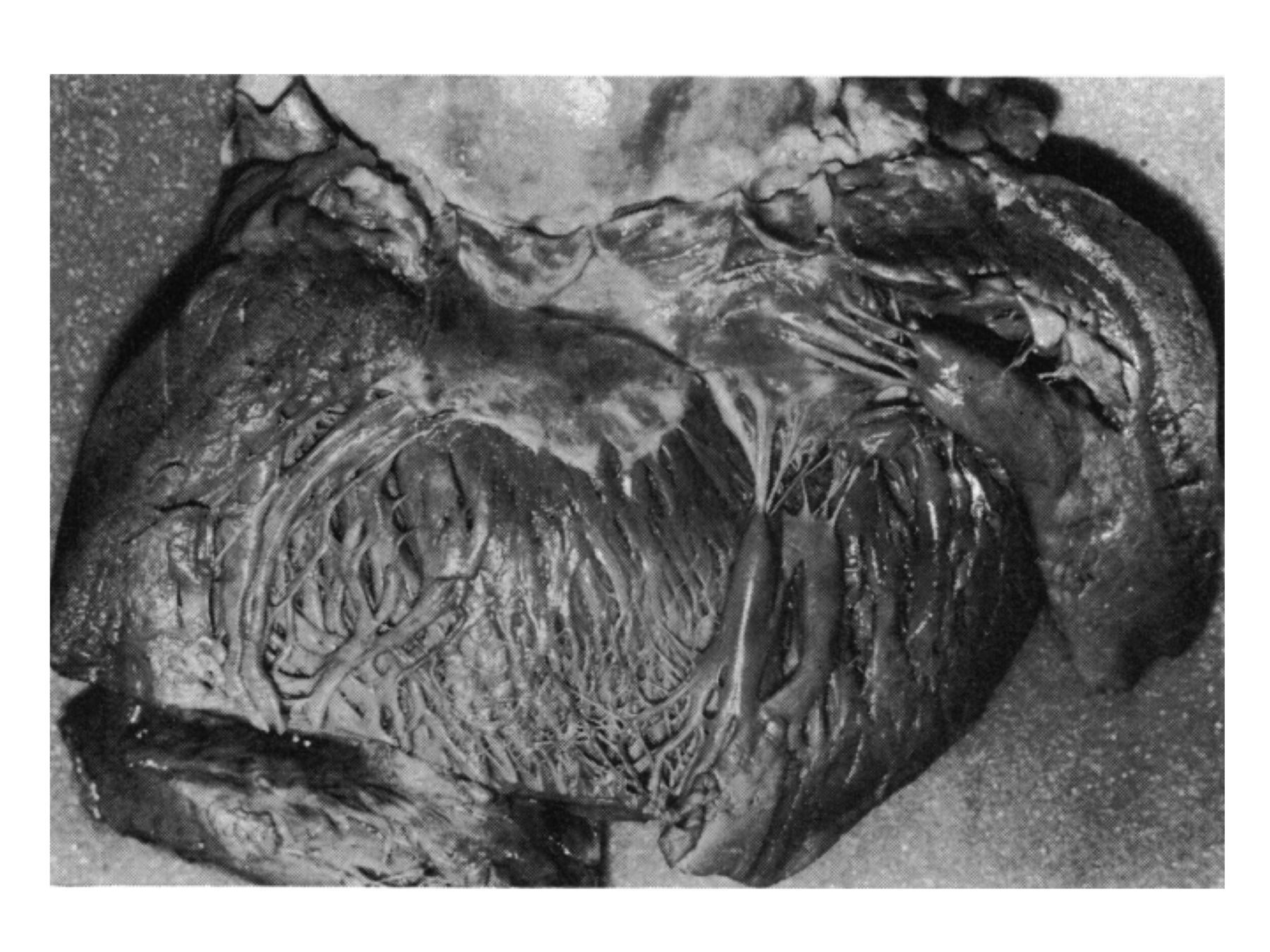 Figure 4
Figure 4 Opened left ventricle from a 61-year-old woman dying suddenly and unexpectedly. Postmortem examination showed a greatly hypertrophied heart, weighing 700 g. The entire left ventricular myocardium is thickened and the cavity slightly dilated. The characteristic subaortic band and its relation to the anterior mitral cusp are well seen. Patchy white thickening of the rough zone of the cusp is also present. Reproduced with permission from Pomerance and Davies58
The pathophysiological substrate for SCD has been well characterised at whole organ and single cell levels.43 44 Notably, it was assumed that many episodes of cardiac arrest with ventricular fibrillation were preceded by brief runs of ventricular tachycardia or sudden-onset atrial fibrillation, but data obtained from implantable cardioverter-defibrillators (ICDs) have shown that ventricular fibrillation can often occur straight from sinus rhythm, highlighting the degree of myocardial vulnerability in HCM.45
Numerous studies have shown that ICDs are highly effective in terminating life-threatening ventricular arrhythmia in patients with HCM. However, the key challenge has always been to identify the small minority of subjects who are at sufficiently high risk to warrant prophylactic treatment with an ICD.46 Various clinical parameters have been proposed as markers of increased risk of SCD,47 and for many years patients were selected for treatment with ICDs based on the presence of one or more such clinical markers. In 2014, the European Society of Cardiology published a risk algorithm based on a multicentre, retrospective cohort study of 3675 patients in which risk factors independently associated with SCD on multivariable analysis were used to construct a clinical decision tool (HCM RISK-SCD) that provides individualised risk estimates. In an independent international validation study, HCM RISK-SCD performed well, with a number needed to treat of 13 in order to save one life, and displayed a markedly higher discrimination than previous algorithms.48 In the latest 2020 American College of Cardiology/American Heart Association guidelines,49 reliance on a simple summation method persists, but there is new emphasis on shared decision making between the healthcare team and the patient. The aim is to deliberately use informed discussion of the strength of the risk factors identified and the level of risk deemed acceptable to make a decision on ICD implantation; HCM RISK-SCD is acknowledged as a key element of this informed consent process. In terms of deciding on the type of ICD being offered, it is relevant to consider the potential for device complications as well as the patient’s age.50 In younger patients, subcutaneous ICDs offer the distinct advantage of minimising the risk of cardiac device-related infective endocarditis.
The future: looking towards novel therapiesDespite improvements in risk stratification and ICD technologies, there remain important unmet needs in the treatment of progressive heart failure. Excitingly, identification of the underlying pathogenic gene variant offers the realistic prospect of therapies that correct or mitigate the effect of causative mutations. Various approaches are under investigation, including RNA-based therapies and gene replacement or editing, and the potential for aetiology-focused therapy has been demonstrated with the use of mavacamten, a first-in-class small-molecule, selective inhibitor of cardiac myosin ATPase, which targets myosin-actin cross-bridge cycling. In the phase III EXPLORER-HCM study, mavacamten significantly improved exercise capacity and symptoms in patients with obstructive HCM at week 30 compared with placebo,51 and trials are now underway in patients with non-obstructive disease with this and similar agents. There is also renewed interest in the repurposing of existing therapeutic compounds, all within the context of randomised clinical controlled trials.52
The changing face of HCMPhysicians in clinic today recognise a much wider spectrum of disease,53 with far more heterogeneity than the original entity described by Teare 60 years ago. Although early studies of patients with HCM from specialist centres suggested a ‘positive’ gene status in up to two-thirds, recent large-scale registry data have clarified that individuals with a negative gene panel test represent the majority.54 Multicentre registries also provide a growing realisation that prognosis tends to be better in sarcomere variant-negative HCM, with lower risks of complications such as heart failure, sudden death, atrial fibrillation and stroke.55 Similarly, familial recurrence risk is lower, and rather than presenting in early adulthood the disease often arises in middle to late age. Apical HCM and paradoxically the presence of left ventricular outflow tract obstruction (once considered a hallmark feature of HCM) are both more common in sarcomere-negative disease.54 The explanation for these observations has recently been provided by a genome-wide association analysis, which suggests that sarcomere-negative HCM is a complex, polygenic trait,56 in which other common cardiovascular traits such as diastolic hypertension may cause disproportionate hypertrophy in genetically susceptible individuals. A personalised approach will be needed involving consideration of a patient’s gene status when assessing the future utility of novel therapies.
ConclusionSince Teare provided us with a prototype for studying the genetics of cardiac disease more than 60 years ago, there has been a relentless pursuit by investigators to better characterise this disease and our understanding continues to evolve. In the modern era, there is already an opportunity to diagnose HCM at a preclinical stage and to provide evidence-based individualised assessments of risk and personalised treatment strategies. For the future, with the advent of RNA-based and gene modification therapies, there is a realistic potential to halt or even prevent the development of disease.
Ethics statementsPatient consent for publicationEthics approvalThis study does not involve human participants.
留言 (0)