
記住我
Astrocytes and microglia, both types of glial cells, are key cells in the central nervous system (CNS), maintaining homeostasis and supporting the function of neurons (Figure 1). The communication between neurons and the glial cells is well recognized. However, crosstalk between astrocytes and microglia is also an essential feature of a healthy CNS, and the breakdown of this communication may be an important mechanism in neurodegenerative disorders. The study of glial communication in humans remains a challenge due to the complexity, access, and function of the CNS. However, tools have been developed in an attempt to overcome these difficulties, with some of them specifically designed to explore glial cells.
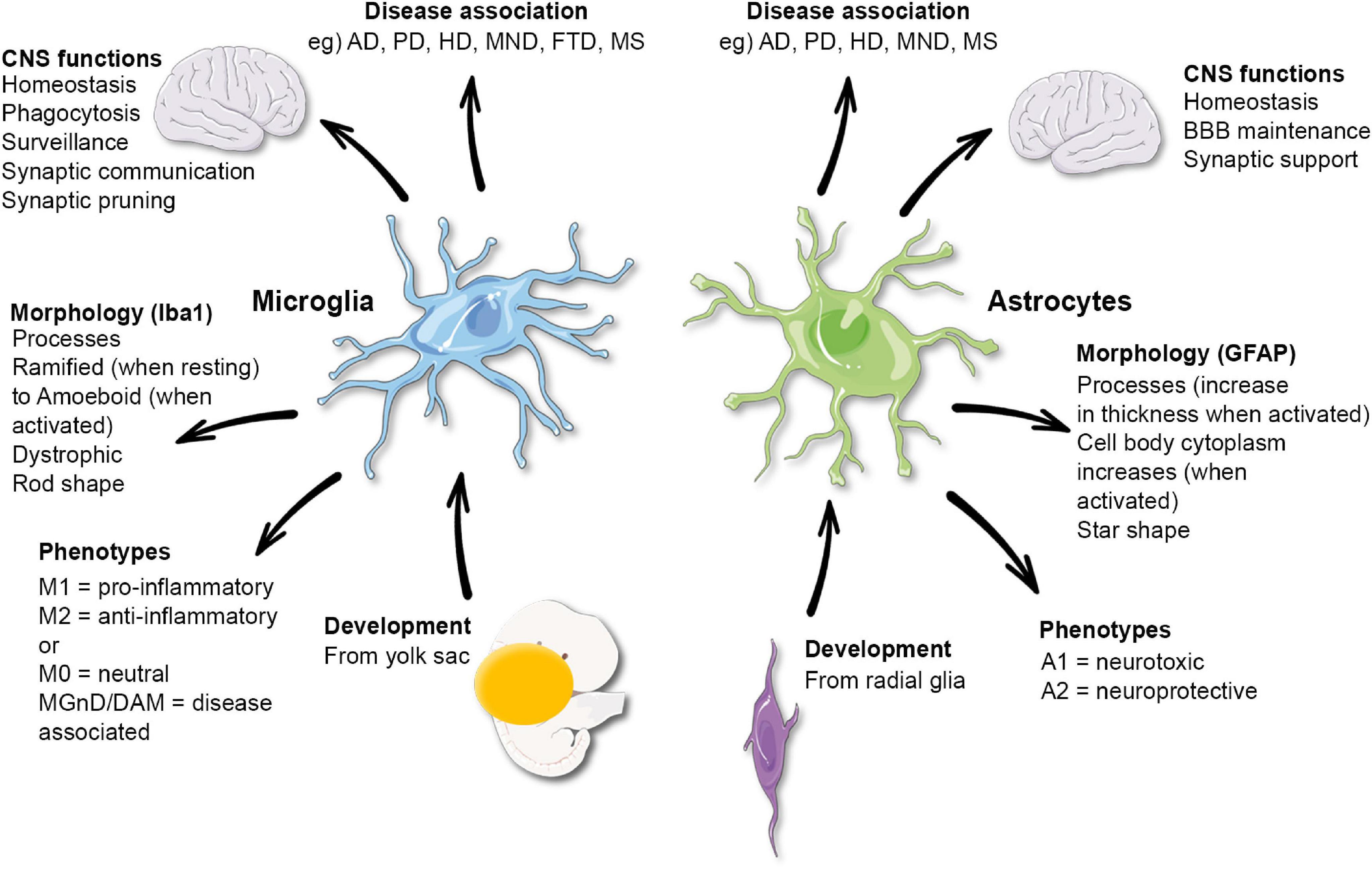
Figure 1. Functional and morphological characteristics of microglia and astrocytes. AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; MND, motor neuron disease; FTD, frontotemporal dementia; MS, multiple sclerosis. Microglia phenotypes have been disputed and may not reflect their functions.
Here we review examples in the literature of the current understanding of microglia and astrocytes functions and their communication in the human brain that has been gained using some of these tools, with a focus on Alzheimer’s disease (AD) and other neurodegenerative diseases.
The research discussed here will include genetic and transcriptomic studies which evaluate genes and proteins expressed by glia and their relevance to disease. The functional studies reviewed look at cultured human cells using primary isolated glial cells, or cells derived from human induced pluripotent stem cells (iPSC). The stem cell model systems have been generated to closely mimic the human environment whilst allowing cellular manipulation to improve our understanding of their functions. For these characteristics, iPSCs have become the most utilized cell system in neurodegenerative disease (Table 1), used individually or in co- or tri-culture systems to investigate cell cooperation and coordination in what is known as the cerebral organoid.
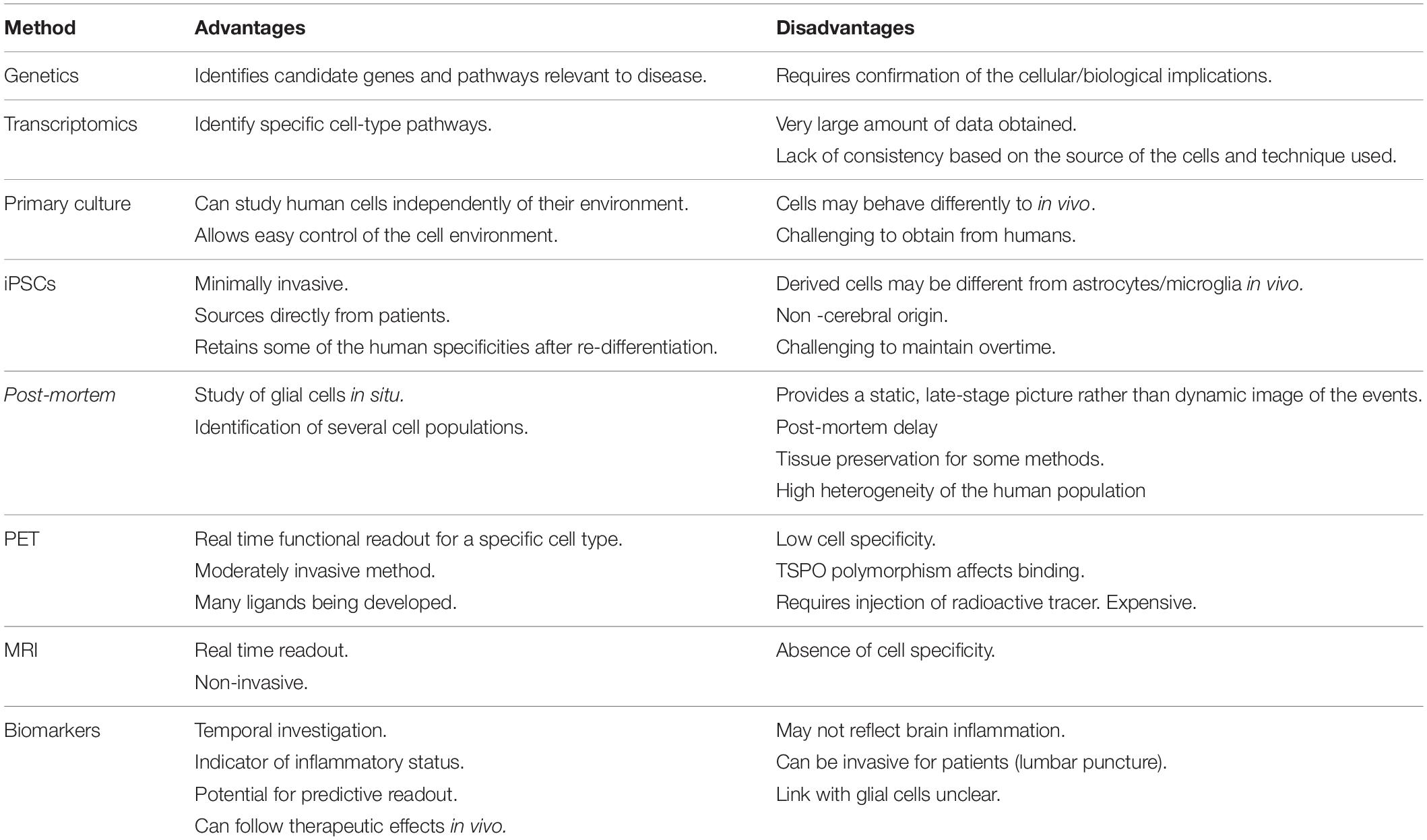
Table 1. Current methods available to study human glial cells.
Other studies have explored astrocytes and microglia in situ in human brain tissue, through post-mortem immunohistochemical studies to examine cells at a specific time in their real environment. Finally, recent non-invasive techniques will be also discussed which assess microglia/astrocyte activation in patients during life, allowing temporal investigation of their activated status via the use of brain imaging and fluid biomarkers (Table 1).
An Overview MicrogliaMicroglia are the resident immune cells of the CNS, accounting for 10% of cells (Salter and Stevens, 2017). They are derived from the yolk sac (Ginhoux et al., 2010), and during brain development regulate neurogenesis (Cunningham et al., 2013), promote neuronal survival (Ueno et al., 2013) and participate in synaptic pruning, to ensure appropriate neuronal connections are made and brain maturation occurs (Schafer et al., 2012). Throughout adult life, they continue to interact with their environment, contributing to synaptic communication and ensuring cerebral homeostasis is maintained within the brain by constantly surveying the surrounding parenchyma with their finger-like processes (Boche et al., 2013). They have recently been defined as a multifunctional housekeeping cell type (Cserép et al., 2021). However, the exact function of microglia is still somewhat unclear in human neurodegenerative disease, but evidence supports a role for these cells in conditions such as AD (Franco-Bocanegra et al., 2019b), Parkinson’s disease (PD) (Lecours et al., 2018), and frontotemporal dementia (FTD) (Hartnell et al., 2021).
Microglia have been considered to exist in two states referred to as ‘resting’ or ‘activated.’ ‘Resting’ microglia were classed as those that are highly ramified in their morphology with processes allowing them to scan the microenvironment for foreign bodies and neuronal injury (Boche et al., 2013). The term ‘resting’ was not accurate as the ramified microglia are highly dynamic cells, as observed in the rodent brain (Nimmerjahn et al., 2005). In the context of cerebral homeostatic changes (e.g., due to infection, damage, or disease), the cells become ‘activated’ and undergo morphological changes that include enlarged cell bodies with thickening and shortening of their processes, leading to a more amoeboid-like morphology and migration toward the “anomaly” (Boche et al., 2013; Franco-Bocanegra et al., 2021). In addition, the morphological modifications are associated with the expression of inflammatory molecules in response to the homeostatic disturbance of the micro-environment (Hanisch, 2002). Two types of microglial activation status were initially considered and named M1 and M2. M1 was referred to as classic activation where the cells expressed pro-inflammatory cytokines, targeted foreign bodies, exhibiting antigen presentation markers and performing phagocytosis (Mosser and Edwards, 2008). The M2 status was known as alternate activation/wound healing. Again, microglia in the M2 profile performed phagocytosis but also promoted changes in the extracellular matrix as a healing process and the profile was associated with the expression of anti-inflammatory cytokines (Edwards et al., 2006). This categorization, which was derived from the macrophages in experimental conditions, is now disputed. Indeed, several microglial populations have been detected cohabiting within the same brain, with specific disease-associated microglial profiles identified in experimental models (Boche and Gordon, 2021) and conditions such as AD, associated with a mix of pro and anti-inflammatory molecules expressed in the brain (Rakic et al., 2018; Franco-Bocanegra et al., 2019a). However, consensus on the number and functions of microglial populations and the presence of disease-specific profiles as identified in models remains to be established in humans.
AstrocytesAstrocytes are a subset of glial cells which contribute to the maintenance and regulation of neuronal function. These star-shaped cells make up between 17 and 61% of the cells in the human brain, depending on the area (von Bartheld et al., 2016). Astrocytes have several functions within the healthy CNS including neurotransmitter cycling, metabolic support of neurons and maintenance of the blood–brain barrier (via their roles in neuro-vascular coupling) (Sofroniew and Vinters, 2010).
Our current knowledge of astrocytes is mostly sourced from studies in rodents; however, as with microglia, human astrocytes differ quite markedly from those of rodents. For example, differences were observed in calcium signaling functions and transcriptome readouts between human and mouse astrocytes (Zhang et al., 2016). There are several subclasses of astrocytes (see Figure 2) the most numerous of which are protoplasmic astrocytes, found in all mammals. These have a stellate morphology and inhabit their own distinct domain in layers II–VI of the gray matter never overlapping other astrocytes. However, in humans, these astrocytes are three times larger with more than ten times the number of projections than those of rodents (Oberheim et al., 2009). There are also two distinct subtypes of astrocytes found only in primates and humans which reside in either layer I or layer VI and reach projections down (or up) into the other cortical layers (Oberheim et al., 2006). These interlaminar astrocytes have high expression of CD44, GFAP (glial fibrillary astrocytic protein) and S100B (S100 calcium-binding protein B), but low expression of glutamate processing markers excitatory amino acid transporter (EAAT)- 1 and EAAT2 and Glutamate Synthetase (Sosunov et al., 2014). Similar transcriptional properties were also seen in the white matter “fibrous astrocytes.”
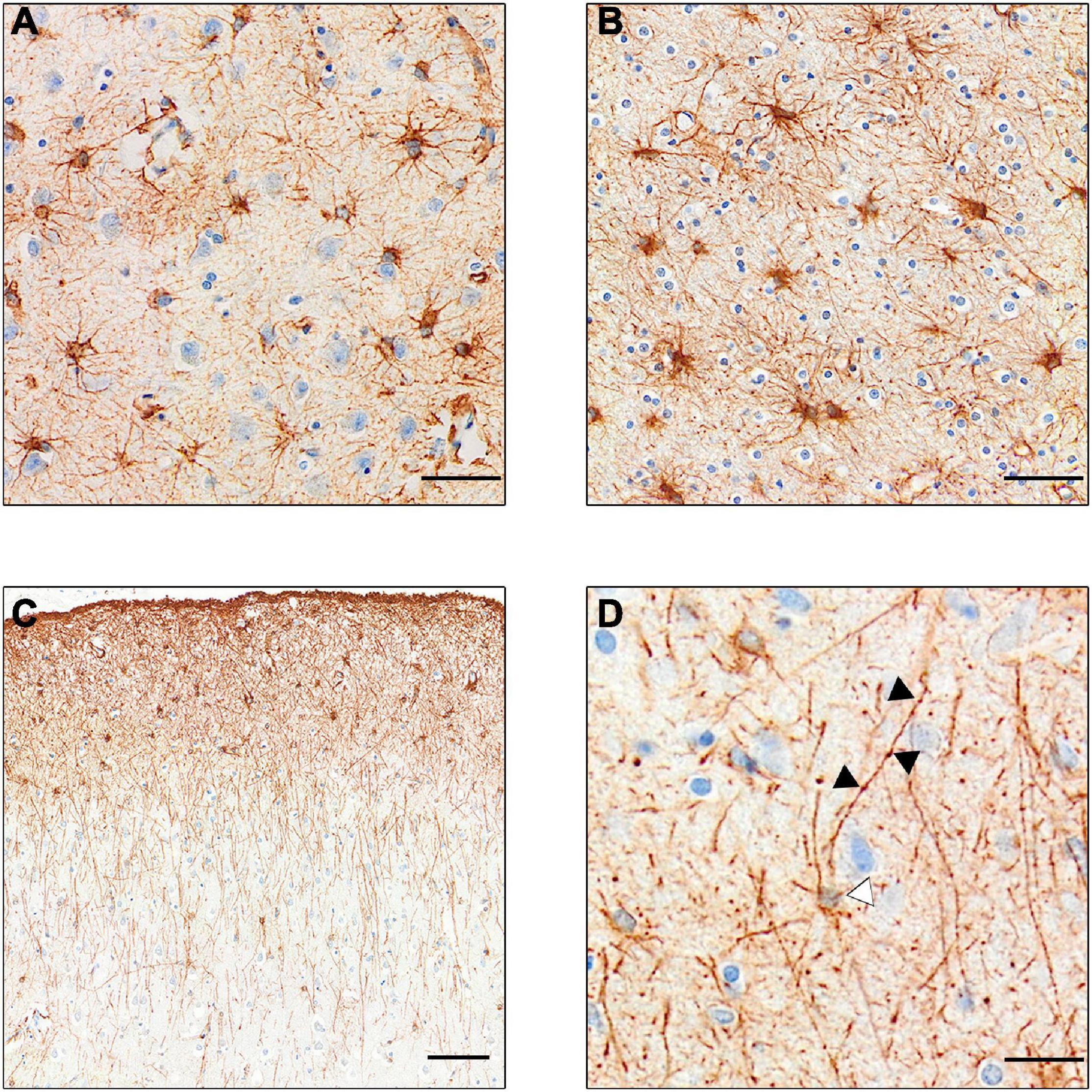
Figure 2. GFAP+ stained astrocyte subtypes. (A) Protoplasmic astrocytes of the gray matter. Note the lighter staining, showing less expression of GFAP. (B) Darker stained fibrous astrocytes of the white matter. (C) Intralaminar astrocytes stretching process from layer I–II to layers III and IV. (D) Varicose projection astrocyte. Black arrows point to varicosities on the straight primary process. White arrow points to faintly stained cell body. [Scale bar (A,B) = 50 μm, (C) = 100 μm, (D) = 25 μm].
Interest has increased in the role of astrocytes during disease, particularly in their participation in the neuroinflammatory processes. Changes in astrocyte morphology, function and abundance (termed “astrogliosis”) have been observed in human neurodegenerative disorders (Pekny and Pekna, 2014). However, many studies used GFAP to monitor astrocyte proliferation, and it is argued that the increased expression may be related to upregulation in individual cells rather than astrocytic proliferation (Perez-Nievas and Serrano-Pozo, 2018), as observed in a study using the proliferating cell marker PCNA (Marlatt et al., 2014). Furthermore, studies using cell counts of astrocytes – through Nissl staining (Pelvig et al., 2003) or constitutive astrocyte markers in combination with GFAP (Serrano-Pozo et al., 2013a) – showed no difference in cell number in AD compared to control brains. Examples of GFAP in a control brain and a brain with Pick’s disease are shown in Figure 3.
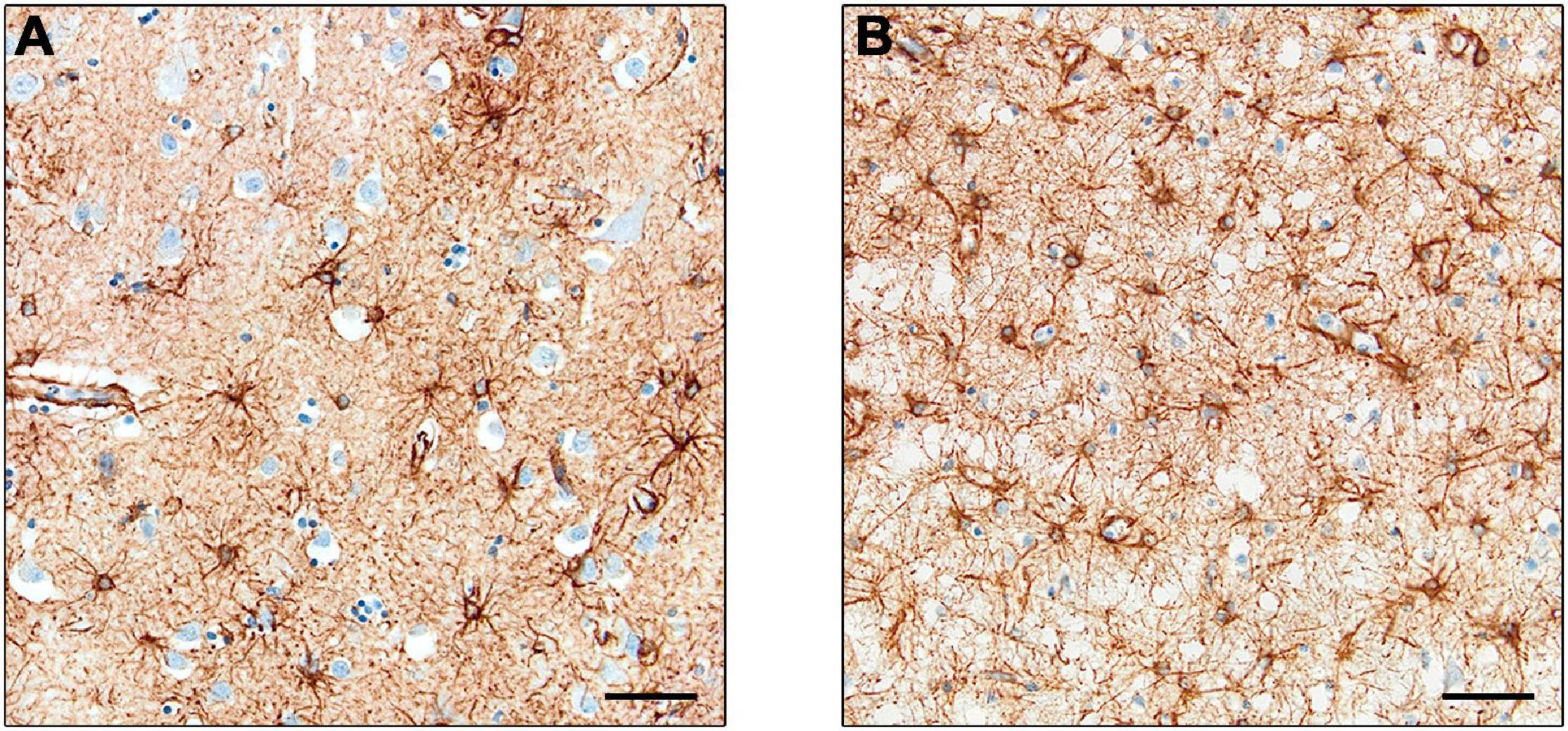
Figure 3. Expression of GFAP+ astrocytes. Astrocytes in control cases (A) and Pick’s disease cases (B), with the latter showing darker staining and greater cell number. Scale bars = 50 μm.
Further to their increased GFAP expression during neuroinflammation, astrocytes take on a neurotoxic phenotype (A1) (Zamanian et al., 2012) which loses neuroprotective abilities, leading to neuronal death. This A1 phenotype was revealed in AD, Huntington’s disease (HD), motor neuron disease (MND), and PD (Liddelow et al., 2017). However, the idea that astrocytes become polarized to an A1 (neurotoxic) or A2 (neuroprotective) phenotype upon activation from different signals issued from microglia may be misleading. As with the previous M1/M2 distinction in microglia (from which the field has moved on) (Ransohoff, 2016), it seems that despite the discovery of a 12 gene signature of A1 astrocytes (Liddelow et al., 2017), intermediate or divergent transcriptomic states might exist and even co-exist within the same brain (Escartin et al., 2021). Other studies have also cast doubt on the polarization of the astrocytic phenotype, showing that the transcriptomic signature of disease-related astrocytes in AD (Grubman et al., 2019; Zhou et al., 2020) or HD (Al-Dalahmah et al., 2020) did not overlap with the one initially described by Liddelow et al. (2017).
A phagocytic role for astrocytes has also be identified in the humans. In AD, reactive astrocytes via their processes are known to form clusters around amyloid plaques and APP-containing dystrophic neurites (Gomez-Arboledas et al., 2018). However, the number of astrocyte processes correlated with the amount of plaque-associated neurites rather than plaque size, suggesting that they are responding to neuronal damage rather than the plaques per se (Serrano-Pozo et al., 2013b). Using immunofluorescence, it was shown that GFAP+ processes closely contact APP-containing neurites. Using transmission electron microscopy, they observed that the astrocytes-neurites association involved astrocytic phagocytosis with dystrophic neurites internalized by reactive astrocytes (Gomez-Arboledas et al., 2018). This supports that a dysfunction in astrocytes’ ability to carry out phagocytosis may contribute to the progression of AD.
Genetics and Transcriptomic Studies MicrogliaGenetic data has been invaluable in providing a greater understanding of microglia and their function in neurological conditions. Being derived from the early erythro-myeloid progenitors in the yolk sac (Ginhoux et al., 2010), many genes contribute to their differentiation and role as immune cells within the CNS (Wurm et al., 2021). As aging occurs, microglial gene expression changes and this might potentially explain why aging is the major contributor to disease risk in neurodegenerative conditions (Olah et al., 2020).
Genome-wide association studies (GWAS) allow the identification of inherited genetic variants associated with disease risk by testing an extremely large data set for important genetic information. It was well established that the APOE (apolipoprotein E) genotype was, with aging, an important risk factor in developing AD (Saunders et al., 1993). There is a protective APOE allele (ε2), a neutral allele (ε3), and a risk allele (ε4). An individual who is homozygous for the ε4 allele has an 8-12 times greater chance of developing AD (Genin et al., 2011; Serrano-Pozo et al., 2015). Twenty years later, one GWAS study identified 11 novel genes associated with the risk of AD. These included HLA-DRB5–DRB1, SORL1, PTK2B, SLC24A4, ZCWPW1, CELF1, CASS4, and FERMT2 genes (Lambert et al., 2013), highlighting the involvement of several immune genes. Other GWAS studies have then focused on the association between microglial genes and AD and found that TREM2, CD33, CR1, and PLGC2 were all significant risk factors for the disease and that they may alter the functionality of microglia (Malik et al., 2015). Of note, from all GWAS studies in AD, the APOE gene was still the strongest risk factor identified for this disease. Triggering receptor expressed on myeloid cells 2 (TREM2) is of particular interest as the identified genetic variant confers the same magnitude of genetic risk as APOE (Guerreiro et al., 2013; Jonsson et al., 2013). However, the TREM2 variant is present in 1% of the population when the APOE ε4 genotype associated with AD is expressed by 30% of the population. There is evidence for upregulation of TREM2 in AD (Perez et al., 2017), but its exact role in this disease and its presence in human microglia remains to be confirmed (Fahrenhold et al., 2018). GWAS studies are crucial for discovering genetic risk factors linked to disease, but they do not show the whole picture of which protein, cell or process is involved. Therefore, to determine the function and role of the gene/protein in particular circumstances, other tools and techniques are usually necessary.
The development of the single-cell RNA sequencing (scRNAseq) method has allowed the identification of different microglial populations based on genetic expression. This is a high-throughput technology (Svensson et al., 2018) that can distinguish between microglia and other myeloid cell populations (e.g., perivascular macrophages) using specific microglial proteins such as the transmembrane protein 119 (TMEM119) (Bennett et al., 2016). This method, in both mouse models and human brain samples, has re-emphasized that microglial genes are highly associated with neurodegenerative disease (Keren-Shaul et al., 2017; Olah et al., 2020). A study using scRNAseq on human brain tissue including mild cognitive impairment (MCI), AD or epilepsy patients, identified 9 different microglial subpopulations with different functionalities, including homeostasis, antigen presentation and proliferation. Four of these subpopulations were similar to the disease-associated microglia transcriptomic profiles defined in experimental models (Olah et al., 2020). Another human study identified seven different populations and did not find a difference in the gene expression profiles of microglia, suggesting an absence of specific AD-related microglial populations (Alsema et al., 2020). The discrepancy between studies emphasizes the challenges associated with this technology when used in the human brain (Boche and Gordon, 2021), partly due to the small number of microglia and the need to pool several brains together leading to conditions being mixed. It also highlights the need to confirm the experimental findings in humans to advance our understanding of microglia.
Single-cell RNA sequencing has more recently challenged the microglia activation classification groups, M1 and M2, with results showing that these may be an oversimplification of the roles of microglia. Evidence suggests a progressive switch in gene expression that causes microglia to become activated, rather than a distinct on or off mechanism. Also, it has been shown that the progressive switch may be related to the proximity of Aβ plaques in the context of AD (Keren-Shaul et al., 2017). In experimental models, transcriptomics identified two main gene clusters associated with microglia, these being a homeostatic cluster given the term ‘M0,’ defined by P2ry12, Tmem119, Cx3Cr1, and an inflammatory cluster termed ‘MGnD’ defined by Ccl2, Csf1, and Apoe. The same study also reported that TREM2 induces the APOE pathway which then causes a switch between M0 and MGnD microglial phenotypes, and thus targeting this pathway could reinstate the homeostatic nature of microglia in AD (Krasemann et al., 2017). Some of the microglial phenotypes described from mouse brains have been confirmed in humans (Boche and Gordon, 2021).
Lastly, single nucleus RNA sequencing (snRNAseq) has been developed as an alternative method to scRNAseq to explore transcriptomics in tissue where it is difficult to extract cells and keep them intact. It also has the advantage of being performed on frozen rather than fresh tissue and thus can make use of archived, well-characterized cases. Using this methodology on two brains, a total of five microglial clusters were identified with the presence of microglia genes highly expressed in some of the clusters such as the homeostatic gene P2Y12 in and C1QA. The authors reported similar findings on microglia isolated from fresh tissue from the same donors and concluded this to be proof of the relevance of the snRNAseq method to study microglial genes (Gerrits et al., 2020). Indeed, nuclei isolated from human microglia can provide a substantial genetic map to investigate important microglial genes. However, this has been challenged by another study also comparing scRNAseq and snRNAseq on microglia isolated from frozen biopsies. They observed that genes associated with microglial activation identified in scRNAseq (e.g., APOE, SPP1, and CD74) were depleted in snRNAseq (Thrupp et al., 2020), implying a low sensitivity of the method and that microglial nuclei may not recapitulate the whole picture of microglial activation. The different studies clearly demonstrated the challenges associated with the study of microglia in humans.
AstrocytesAs with microglia, APOE is also an important gene when it comes to astrocytes in the context of AD. Astrocytes are the primary APOE expressing cells of the brain. In humans, APOE ε4 carriers had similar numbers of GFAP+ astrocytes (either reactive or resting) to other APOE genotypes (Serrano-Pozo et al., 2011). Whilst similar in number, their function may be altered, as in astrocytes derived from human iPSCs, an APOE ε4/ε4 genotype was found to cause a loss of neurotrophic function (Zhao et al., 2017).
To uncover other genes in astrocytes that may be involved in the pathological process in AD, transcriptomic studies compared astrocyte genomes between control and AD brains. Laser capture microdissection was utilized to isolate astrocytes from post-mortem human tissue for their transcriptomes to be analyzed using gene microarrays. This technique has allowed the identification of several astrocytic genes including the mitochondrial genes PITRM1-AS1 (pitrilysin metallopeptidase 1 antisense RNA1), an antisense copy of a gene known to degrade Aβ (Falkevall et al., 2006), NDUFA4L2 (NADH dehydrogenase 1 alpha subcomplex, 4-like 2) which inhibits respiratory complex 1 in mitochondria, and FASKD2 (fast kinase domain-containing protein 2) involved in regulating apoptosis. Other immune-related genes found to be different in AD astrocytes were C3 (complement component 3), CLU (Clusterin) and CD74 (cluster of differentiation 74) (Sekar et al., 2015). Another study also reported changes in genes across several functional pathways between early and late-stage AD. The main differences were detected between the Braak stages 1–II vs. Braak stages V–VI and were related to reductions in signaling pathways related to insulin, MAPK (mitogen-activated protein kinase), PI3K (phosphatidylinositol 3-kinase), and PKB (protein kinase B). Furthermore, they revealed that many pathways in astrocytes were also modified with respect to the APOE genotype (Simpson et al., 2011).
Human Primary Cell Cultures and Induced Pluripotent Stem CellsCell culture has been a commonly used method to study glial cells and allows the control of the physiochemical environment of the cell. However, it was argued that the representation of the native/physiological environment of cells was limited in single-cell culture model systems though there are ways of mimicking in vivo systems in this in vitro method. Several glial cell lines are commercially available, some of them derived from human cells, but for cell lines to acquire the necessary properties to grow in vitro, there is a loss of their specificity.
The culture of primary glial cells has the advantage of being prepared from cells isolated directly from the brain and hence provide more relevant findings than cell lines. These cells are often sourced from rodent pups, and are not representative of differentiated/mature cells and thus require to be highly manipulated to mimic neurodegenerative diseases associated with aging. In addition, human microglia and astrocytes are known to be significantly distinct from those of the rodents (Oberheim et al., 2009; Franco Bocanegra et al., 2018). Therefore, primary human cell culture can provide avenues to explore human cell physiology in health and disease, although with similar limitations as for cell lines, namely its simplicity and disconnection from the original cell environment. Furthermore, primary cell culture from humans is a particular challenge, as it requires either fetal cells (that may not represent those in chronic disease), or cells collected from adults during neurosurgical operations (that might not be relevant to normal physiology or neurodegenerative diseases), or cells isolated from post-mortem brains. Overall, primary human cells are extremely challenging to obtain and involve complex ethical considerations.
Induced pluripotent stem cells are a more accessible model in which to study the behavior of human cells. Cells from the skin and blood (Lowry et al., 2008) are sourced from an individual and reprogrammed into embryonic stem cell-like cells (known as iPSC cells) using the Yamanaka factors and have been generated from a variety of differentiated cells (Takahashi and Yamanaka, 2006). iPSCs can be re-differentiated into any cell type and iPSC-based methods are now widely used to study the physiopathology of disease or as platforms in drug discovery with iPSCs banks being established.
Microglial Cell Culture ModelsMicroglia derived from human iPSCs have been generated to mimic physiological microglia (sourced from healthy donors) or disease microglia (provided by patients). Microglial cells derived from healthy human iPSCs were able to phagocytose foreign particles, as previously observed in cell cultures, but most importantly expressed microglial-specific markers such as TMEM119. Furthermore, when microglial iPSCs were co-cultured with neurons, the microglial signature was enhanced and the microglial cells presented a ‘surveying behavior’ similar to the process in physiological conditions. Lastly, when damage was caused to the cell co-culture, the microglia reacted within minutes to these stimuli by extending their processes to the site of injury (Muffat et al., 2016), starting the inflammatory process as expected (Nimmerjahn et al., 2005; Franco-Bocanegra et al., 2019b). One study examined the consequences of the main AD risk factor APOE ε4 on microglia. Interestingly, microglia associated with this allele revealed a different morphology compared to those associated with the APOE ε3 allele. APOE ε4 microglia presented decreased process length and were fewer in number (Lin et al., 2018), which might be associated with decreased phagocytic capabilities, specifically phagocytosis of Aβ. Furthermore, with the use of the CRISPR/Cas9 system, iPSCs with the APOE ε4 allele were converted to the APOE ε3 allele and this consequently caused a reduction in AD pathology (Lin et al., 2018), suggesting that the role of APOE as a risk factor for AD might be triggered by microglia. Interestingly, a study observing the effect of TREM2 mutations on microglia using human iPSCs, found that the missense mutations of this gene caused microglia derived from these donors to express less TREM2. This initiated a reduction in phagocytic capabilities of the human iPSC derived microglia, suggesting that TREM2 missense mutations cause dysfunction in microglia (Garcia-Reitboeck et al., 2018). Furthermore, using iPSCs derived from individuals with a TREM2 mutation that causes FTD, it was discovered that the mutant form of TREM2 could not be processed by microglia, staying in its immature form, and was not properly relocated to the plasma membrane of the cells. However, it was shown that the microglia with this TREM2 mutation could still respond to inflammatory stimuli (Brownjohn et al., 2018). The recent combination of iPSCs based on identified risk factors has highlighted potential microglial mechanisms in AD and other dementias. One study was able to demonstrate that human iPSC microglia recapitulated the ability to phagocytose Aβ and tau both in vivo and in vitro. The cells were also able to release cytokines when exposed to inflammatory stimuli and underwent the same genetic changes from said stimuli as physiological microglia would behave in human brain (Abud et al., 2017; Garcia-Leon et al., 2020).
To attempt to closely recapitulate the microenvironment of the human brain, multiple cerebral cell types have been cultured together. One study developed a co-culture of embryonic iPSC-derived macrophages with iPSC-derived cortical neurons. In this model, macrophage cells matured into microglia, expressed microglia-specific markers such as P2Y purinoreceptor 12 (P2RY12) and MER proto-oncogene tyrosine kinase (MERTK), portrayed their phagocytic function, and were ramified (Haenseler et al., 2017). It was also observed that the iPSC-derived microglia were able to become activated, taking on an amoeboid morphology and releasing inflammatory products. The co-culture was stable for many weeks and microglia expressed many genes associated with AD, such as TREM2, APOE, APP, and PARK15 (Haenseler et al., 2017), making this an effective tool for studying microglia in disease.
Using the opportunity to access brain tissue at the time of the post-mortem examination, isolated microglia expressed constitutive microglial markers such as CD11b, CD32, CD64, CD68, and HLA-DR. Cells sourced from AD brains also presented elevated pro-inflammatory molecules including cytokines interleukin 6 (IL6), tumor necrosis factor-alpha (TNFα), CCL3, CCL4, CXCL8, and M-CSF (macrophage colony-stimulating factor) suggesting a greater inflammatory response from microglia in AD (Lue et al., 2001). A study showed that it was possible to distinguish microglia (CD11b++CD45dim) from macrophages (CD11b++CD45high) using flow cytometry after extraction from post-mortem brain tissue (Melief et al., 2012). Upon LPS stimulation, microglia responded by upregulating inflammatory markers (Melief et al., 2012) as seen in vivo after ‘injury’ occurring in the human brain (Zrzavy et al., 2019). Further analysis of this isolation method has shown the technique to be extremely efficient, yielding up to 450,000 cells per gram of brain tissue (Mizee et al., 2017). Examining the role microglia play in the spread of tau in AD, primary human microglia were isolated from post-mortem disease tissue and analyzed for tau seeds. The study reported that Tau seeds were form of misfolded tau protein and were thought to be the template from which the spread of pathological tau occurs. Interestingly, the isolated microglia contained the tau seeds and were able to further release them into the surrounding media. This suggests that microglia could uptake the tau via phagocytosis with the aim to remove the toxic protein; however, this process does not appear to be sufficient to neutralize the tau seed (Hopp et al., 2018).
It is assumed that as the cells are sourced directly from a donor brain, they may better recapitulate the characteristics of human microglia in vivo than microglia isolated from a rodent brain or human iPSCs cultures. However, this process needs to be improved as the microglial-specific markers were observed to diminish over time in culture (Mizee et al., 2017), making the technique less effective for long-term use without stability improvements and thus less attractive for the study of chronic neurodegenerative conditions.
Astrocytic Cell Culture ModelsAstrocytes can also be successfully derived from human iPSCs, through intermediate-stage neural progenitor cells (NPCs) (Shaltouki et al., 2013; Tcw et al., 2017) or oligodendrocyte progenitor cells (Jiang et al., 2013). Similarly to microglia, astrocytes can be derived from healthy individuals’ iPSCs or generated from patients with known genetic mutations to understand familial diseases.
Deriving astrocytes from the iPSCs of a patient with the PSEN1 M146L familial AD mutation created cells that expressed normal astrocyte markers GFAP, S100B, EAAT1, and Glutamine Synthetase (although cellular localization of the former three markers was altered). These cells were atrophied, with lower surface area and volume than those from the control donor. However, regardless of the presence or absence of the PSEN1 M146L genotype, iPSC-derived astrocytes expressed the chemokines CXCL8 and CCL2, as well as tissue inhibitor of metalloproteinases 2 (TIMP-2) (Jones et al., 2017). The effect of APOE genotypes on iPSC-derived astrocytes was also investigated, with the APOE ε4/ε4 astrocytes showing similar morphologies and expression of the same astrocyte markers and chemokines (with the addition of the upregulation of CCL4 and CCL5) to the PSEN1 M146L astrocytes (Jones et al., 2017). APOE ε4/ε4 astrocytes also showed evidence of increased cholesterol synthesis and release and had impaired clearance of oligomeric Aβ42 (Lin et al., 2018), and were less effective in supporting neurons as illustrated by lower levels of synaptic proteins in the co-culture (Zhao et al., 2017). Exacerbated cytokine expression was also shown in another iPSC study (from PSEN1 ΔE9 mutation patients) which also noted increased Aβ pathology and Ca2+ release from the endoplasmic reticulum of mutant cells (Oksanen et al., 2017). These results may suggest an astrocyte contribution to AD through altered inflammatory protein expression associated with an atrophic morphology.
To understand the activity of human astrocytes, iPSC-derived cells were cultured under different conditions using signaling factors to modify the astrocytes phenotypes to either mature or reactive cells observed in vivo. Incubation with TNFα led to increased expression of CXCL8, CCL5, and lipocalin 2 (LCN2) (Roybon et al., 2013), and co-stimulation with TNFα and IL1β increased CXCL8, CCL5, and LCN2, as well as Complement C3 (a marker of A1 astrocytes) and IL6 (Hyvärinen et al., 2019). Of note, these inflammatory molecules along with the chemokines CXCL10, CCL2, and CXCL8 were also increased in human primary fetal astrocytes after TNFα stimulation (Croitoru-Lamoury et al., 2003; Meeuwsen et al., 2003). Since TNFα is primarily released from activated microglia in the brain, it reinforces the idea that microglial activation has consequences on the astrocyte profiles. To look at astrocytic aging, cells derived from NPCs from young (0.5–3 years) and older (42–56) donors were compared. A more reactive phenotype was noted in older derived astrocytes, which had larger surface areas and showed higher intensities of two intermediate filament proteins, GFAP and vimentin (Gatto et al., 2021).
Microglia/Astrocyte Co-culturesTo mimic the complexity of the human brain, a recent model was developed encompassing human NPCs alongside human microglia and astrocytes (Park et al., 2018). This revolutionary system used a 3D microfluidic approach which allowed the recapitulation of the cells natural microenvironment by creating chemical gradients (Halldorsson et al., 2015). For the first time in vitro, this 3D human tri-culture model was able to represent the kinetics of the pathophysiological mechanism of key features of AD at the different stages of the disease, with the presence of Aβ aggregation, microglial activation and tau accumulation (Park et al., 2018). The increased cytokine levels observed suggested that microglia were being activated in response to and migrated toward Aβ deposition and this was associated with neuronal/neuritic damage. In addition, neurotoxic activity, axonal cleavage and nitric oxide release were observed, all consistent with inflammatory features of human AD (Park et al., 2018).
Post-Mortem StudiesPost-mortem examination remains the gold standard for the diagnosis of neurodegenerative conditions and thus has been essential in emphasizing the role of glial cells in disease. However, this type of study only provides a ‘snapshot’ rather than a temporal profile of the events, usually assessing the state of the brain at the end stage of the disease. Occasionally studies attempted to assess progression by designing cohorts with a spread of neuropathological severity. However, since obtaining post-mortem tissue can be problematic, this is not always possible. Another challenging aspect of working with human brains is the high number of cases required to achieve statistical power and meaningful data due to human variability. This contrasts with animal studies in which data are less varied due to the animals being genetically identical, and thus require a smaller number of animals, but they do not replicate the complexity of human diseases. This emphasizes the need for human comparison, and in the last 20 years, the establishment of brain banks has facilitated the access of human tissue to researchers (Gomez-Nicola and Boche, 2015; Hartnell et al., 2021). This has been associated with the development of antibodies specific to glial proteins, essential to the morphological and functional identification of the cells allowing qualitative and quantitative studies (Magaki et al., 2019) (Table 2). By comparing control and disease brains, the effects of disease on microglia and astrocyte number, morphology, and function have been explored.
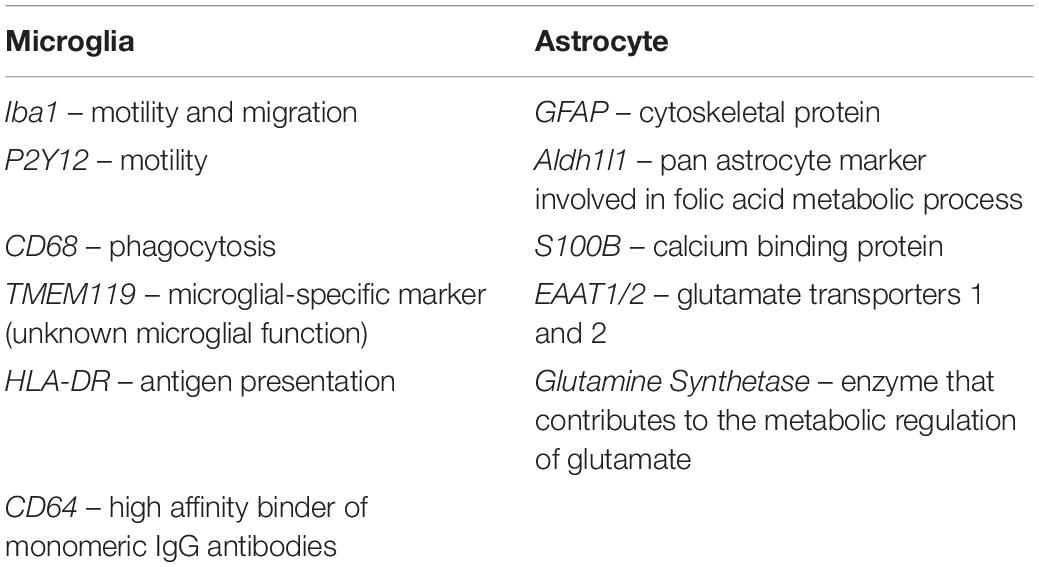
Table 2. Most common microglia and astrocyte immunomarkers and their functions.
Microglial StudiesNumerous post-mortem studies have been performed concerning microglia in AD, and thus a few recent examples will be described here to highlight our current knowledge. For example, a systematic review conducted in 2017 demonstrated that the markers CD68 and major histocompatibility complex (MHC II), acknowledged as markers of microglial activation linked to phagocytosis and antigen-presentation, respectively, were increased, while the expression of CD11b (complement receptor 3) and ionized calcium-binding adapter molecule 1 (Iba1) (Figure 4), associated to microglial motility (Franco-Bocanegra et al., 2019b) were not different between the control and AD cohorts. This was confirmed by quantification of cell counts, which stated the number of microglial cells did not differ between the cohorts, but their activation was increased (Hopperton et al., 2018). This implies that microglia were responding to the disease state by altering their functions (becoming activated) rather than by increasing their numbers. A study investigating microglial activation in the hippocampus during the course of the disease described an abnormal morphology of microglia in the late stage of AD. Microglial morphology was characterized by the presence of fragmented processes (known as dystrophic microglia), a decrease of the cell arborization and of the cell numbers associated with a decrease of the area of surveillance usually monitored by microglia. These features were observed using Iba1 and P2RY12 immunostaining and were consistent with the absence of increased expression of several microglial genes (CD11b, Iba1, TREM2, and CD33). The abnormal morphology was interpreted as microglial degeneration, highlighting the presence of dysfunctional microglia in AD, possibly as the result of the accumulation of phosphorylated tau, the dominant neuropathological hallmark in the hippocampus (Sanchez-Mejias et al., 2016). Of note, both studies reported the absence of increased cell number but the presence of different phenotypes of microglia, differences maybe due to the immunomarkers used, the brain area studied and/or the pathology presents, highlighting the heterogeneity of the microglial populations and of their responses in human AD.
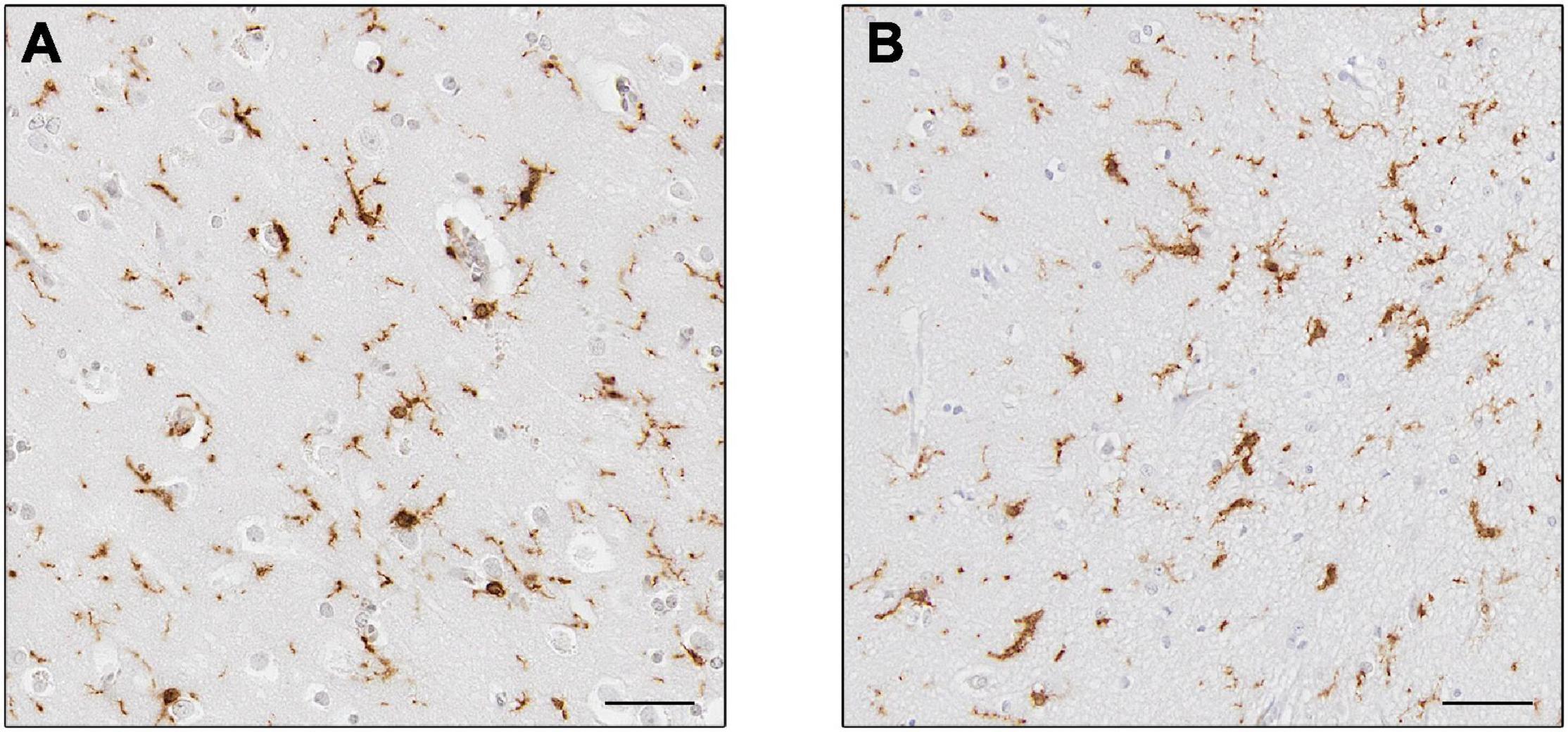
Figure 4. Expression of Iba1+ microglia. Microglia in controls (A) and Alzheimer’s disease (B). Cells in (B) appear to have thicker processes. Scale bars = 50 μm.
Nevertheless, studying individual markers of microglial functions does not capture the variation of microglial phenotypes present in the brain, so one study used a combination of seven microglia/macrophages markers (TMEM119, CD32, CD74, CD163, CD206, P2Y12, and L-Ferritin) and the activated marker (HLA-DR) to establish the functions of microglia in non-pathological post-mortem brain tissue. HLA-DR was highly co-labeled with CD32, CD163, and CD206 (Swanson et al., 2020). CD32 is a Fc gamma receptor (FcγRII) involved in phagocytic capabilities of microglia (Peress et al., 1993), while CD163 (haptoglobin-hemoglobin complex receptor) and CD206 (mannose receptor) (Galea et al., 2005) are associated with the alternate activation profile (Walker and Lue, 2015). This study showed that microglia possess high heterogeneity in post-mortem tissue, based on the high vs. low expression as done in flow cytometer cell sorting. They detected different functional groups including a microglial population defined as Iba1low/L-Ferritinhigh associated with CD74 and HLA-DR expression and defined as dysfunctional based on its overrepresentation in AD, consistent with single-cell transcriptomic studies (Swanson et al., 2020). The use of myeloid markers means that the findings were not able to definitively be accounted for by microglia only, implying the involvement of perivascular macrophages. Indeed, as mentioned by the authors in their study (Swanson et al., 2020), CD206 is specifically expressed by perivascular macrophages (De Picker et al., 2021; Shtaya et al., 2021).
Another study examined the levels of Translocator Protein (TSPO) in post-mortem brains. TSPO has been identified as a positron emission tomography (PET) ligand to assess microglial activation in patients in vivo (discussed further in the imaging section) (Werry et al., 2019). Immunodetection of TSPO showed that AD patients had significantly higher expression of the protein, consistent with the increased signal observed in patients, and interpreted as an increased amount of activated microglia in AD. However, they also reported astrocytes, endothelial cells and smooth muscle cells positive for TSPO staining and an absence of a significant difference in TSPO expression between controls and AD. Therefore, it was concluded that the TSPO function remains to be fully understood (Gui et al., 2020).
A large-scale post-mortem study was conducted on 299 cases, where brain tissue was immunohistochemically analyzed for different microglial phenotypic markers such as CD68 (phagocytosis), HLA-DR (antigen presentation), Iba1 (microglia motility), methionine sulfoxide reductase (MSR-A – phagocytic marker specific to Aβ) and CD64 (FcγRI with high affinity for immunoglobulins). The study revealed that CD68, MSR-A, and CD64 were increased in relation to cognitive decline in AD, whereas Iba1 was decreased in these brains (Minett et al., 2016). This indicated that phagocytic function was increased with worsening AD, highlighting the microglial response to neurodegeneration. Of note, the opposite relationship with Iba1 and cognition indicated that microglia were less motile and thus less able to support neurons and potentially to respond to a further insult as the disease might be developed. Furthermore, the APOE ε4 (risk) allele was associated with CD68, HLA-DR, and CD64 expression, suggesting an exacerbated microglial response in presence of ε4. In contrast, the protective APOE ε2 allele was associated with microglial motility (Iba1), essential to the physiological function of microglia (Minett et al., 2016). This study highlighted the importance of microglial motility in the response to homeostatic changes in keeping the brain healthy. Using the same cohort, TREM2 immunostaining was performed and interestingly, monocytes rather than microglia or perivascular macrophages were immuno-labeled. This was in contrast with the findings in experimental models and may highlight another difference between rodents and human microglia (Fahrenhold et al., 2018).
A limitation of human post-mortem studies compared to animal models is the difficulty investigating changes in microglia after disease manipulation. Therefore, access to post-mortem tissue from patients involved in clinical trials should be a priority. Indeed, access to the brains of AD patients immunized against Aβ has allowed unique observations. For example, exploration of microglial motility was performed using Iba1, P2Y12, CFL1, and Coronin-1A (CORO1A) markers in association with Aβ and tau (Franco-Bocanegra et al., 2019b). No difference in motility markers was detected between controls and AD cases; while in immunized AD cases, the homeostatic markers Iba1 and P2Y12 were both increased. Interestingly, Aβ was associated with Iba1 and P2Y12 in controls, but this association was lost in AD and not restored in the immunized AD cases. This suggested that microglia respond to Aβ deposition in the healthy brain but then the cells became dysfunctional in AD and that immunotherapy did not restore the physiological conditions, despite the increased expression of both markers (Franco-Bocanegra et al., 2019b).
In the last 5 years, numerous microglial markers have been developed as heterogeneity of microglia within the same brain has emerged from transcriptomics and animal models. The development of novel methodologies to use post-mortem tissue such as multiplex staining will bring key information on glial cells in humans. A study investigating human microglia heterogeneity used multiplex staining of 57 different markers to determine if microglia within the same brain can have different phenotypes and whether they were distinguishable from other mononuclear cells. Microglia obtained from healthy donor brains had a distinct phenotype from other myeloid cells, with TMEM119 and P2Y12 being only and highly expressed by microglia (Böttcher et al., 2019). Furthermore, using multiplexed single-cell mass cytometry, microglia from the sub-ventricular zone and thalamus of post-mortem brains had a marked difference in phenotype compared to microglia from other brain regions, with an increase in expression of CD11c, CD45, CD64, CD68, CD195, CX3CR1, and HLA-DR, profile that they proposed reflected an activated microglia phenotype (Böttcher et al., 2019). This re-emphasized the heterogeneity of microglia within the same brain in humans.
Astrocyte StudiesSeveral astrocytic proteins are considered constitutive markers of astrocytes and are used to detect these cells in human tissue. The most commonly used marker is GFAP, a major component of the intermediate filament of the astrocyte cytoskeleton. As such, it highlights extensive astrocytic processes (Waller et al., 2016) throughout the cortex, staining all four subtypes of astrocytes in the human brain (Oberheim et al., 2009) (Figure 2). The increased expression of GFAP is a feature of several neurodegenerative disorders (Pelvig et al., 2003; Schofield et al., 2003) and is thought to relate to reactive astrocytosis (Escartin et al., 2021). However, it is contested as to whether this is the consequence of increased astrocyte number or increased GFAP expression in individual astrocytes (Perez-Nievas and Serrano-Pozo, 2018). S100B and aldehyde dehydrogenase (Aldh1l1) are astrocytic markers restricted to the cell body in humans with the latter preferentially expressed in gray matter astrocytes (Waller et al., 2016). Aldh1l1 and GFAP were increased in AD, but in patients with dementia with Lewy bodies (DLB), only an Aldh1l1 increase was detected (Garcia-Esparcia et al., 2018), highlighting different pathological mechanisms between the 2 most commons types of dementia (rather than a common mechanism) with a different astrocytic contribution.
Functional markers are now often used, mostly related to glutamate processing. EAAT1 and 2 are excitatory amino acid transporters responsible for the uptake of glutamate from the synapse into astrocytes for cycling. These markers stain extensive process trees of stellate astrocyte in the gray matter. Glutamine synthetase is an enzyme in astrocytes that breaks down glutamate into glutamine after uptake through the EAATs. It primarily stains the astrocyte cell bodies but may also stain the somatic end of the processes (Waller et al., 2016). Astrocytic glutamate cycling may play an important role in mediating the disconnection between pathology and dementia symptoms. It has been shown that cognitively normal patients with AD pathology have astrocytes with longer and thicker processes than those with dementia (Kobayashi et al., 2018). Importantly, as well as showing neurodegeneration, AD pathology patients with dementia symptoms also show a reduction in EAAT2 expression (Kobayashi et al., 2018).
Finally, as activated astrocytes possibly exist with neuroprotective and neurotoxic phenotypes, it is important to detect cells of these phenotypes with specific. As C3 was upregulated in only A1 astrocytes (Liddelow et al., 2017), it has been assumed to be a marker of this phenotype, although this simplification is now disputed (Escartin et al., 2021). Nevertheless, the presence of C3+/S100β+ cells was increased in several neurodegenerative diseases including HD, AD, MND, and PD (Liddelow et al., 2017). Another marker, guanylate binding protein 2 (GBP2) is also considered to be indicative of A1 astrocytes. Whilst both markers showed an increase in Creutzfeldt-Jakob disease (CJD) cases, a disease associated with severe astrogliosis, in controls, C3 displayed high non-specific background staining in human tissue, whereas and GBP2 was highly abundant in GFAP+ astrocytes in disease tissue and was completely absent in control tissue (Hartmann et al., 2019). The C3 staining highlights the challenges in staining diffusible proteins compared to proteins located in the cell compartment or at the cell membrane, thus GBP2 seems to be more suitable to identify polarized A1 astrocytes in humans. Of note, the relationship between constitutive and functional markers is important to consider as situations exist where levels of the EAAT2 marker were unaltered, yet the levels of a constitutive maker (Aldh1l1) were changed (Garcia-Esparcia et al., 2018), which may actually represent the proliferation of astrocytes, yet a downregulation of EAAT2 in each astrocyte. Thus, functional alterations in astrocytes may be present in the condition but missed when viewing markers independently.
Imaging StudiesThe most challenging aspect in the study of microglia and astrocytes is that the cells are not visible during life. iPSC-derived cells are taken out of the system in which they act and are usually derived from non-cerebral cells, and cells studied post-mortem are not dynamic. Therefore, methods have been developed to non-invasively assess microglia and astrocyte activity in the human brain through imaging techniques. This can be done via PET imaging that involves the administration of a radioactive PET ligand to patients which will cross the blood–brain barrier and recognize specific receptors in the brain (Boche et al., 2019). These radioactive ligands release gamma rays that are detected by gamma cameras to show the signal of receptor-bound ligands in 3-dimensions. These ligands bind to receptors whose expression is related to the activation of microglia and astrocytes and this can show the neuroinflammatory status of the brain in vivo. Magnetic resonance imaging (MRI) is another method in which neuroinflammation can be detected. Specialized MRI techniques can show the diffusivity of water through tissue and highlight areas of gliosis (where the flow of water is disrupted by neuroinflammation). While MRI can be a good method of visualizing neuroinflammation, PET imaging is the current gold standard as a functional imaging technique to detect the activation of specific cell types.
Microglial ImagingThe first PET ligand to identify microglia and neuroinflammation is the translocator protein (18KDa) (TSPO) ligand, as mentioned above. Initially, this molecule was called peripheral benzodiazepine receptor (PBR) but later changed to suit its structure and function more accurately (Papadopoulos et al., 2006). There are many different TSPO ligands with their efficacy constantly improved in terms of binding capabilities and half-life. The ligand [11C]PK11195 was the first TSPO-PET ligand used to identify microglia with findings showing its binding increased in patients with AD compared to controls (Cagnin et al., 2001). Limitations of PK11195 impeded its use, including short half-life, a high signal to noise ratio meaning that non-specific binding may occur (Werry et al., 2019).
The DPA family of TSPO ligands is a group of second-generation PET ligands which have come to the forefront of microglia research. These are fluorine-based with a longer half-life and a better microglial specificity than the previously used carbon-based ones like PK11195 (Werry et al., 2019). However, the use of the second-generation ligands has been hampered by the binding affinity linked to the single nucleotide polymorphism (SNP) in the TSPO gene with no binding in the presence of the rs6971 SNP, which affects 1/3 of Caucasians (Werry et al., 2019). Consequently, patients have to be screened for TSPO polymorphism and analysis of TSPO binding affinity to correct for this factor. Among these ligands, DPA-714 has shown interesting results in the time course of AD. A study showed that patients with a high initial DPA-714 binding presented a lower level of microglial activation in the later stages of the disease. Whereas, in patients with a low TSPO binding level in early AD, microglial activation was later increased and had worse disease severity (Hamelin et al., 2016). The authors proposed a protective role of microglia early in the disease.
Since, the third generation of TSPO ligands has been designed to counter the effects of the TSPO polymorphism, based on improving the PK111-95. The [11C]ER176 ligand has so far only been tested in healthy controls. This ligand has been developed based on the incorporation of fluorine-18 to increase the stability and thus facilitate the use of TSPO-ligands in the clinical environment, but further work is required to assess its benefit in a clinical environment.
Translocator Protein is not the only target for microglia PET imaging. As it is currently unknown whether TSPO shows a pro- or anti-inflammatory microglial phenotype, other ligands are being developed in order to explore the inflammatory profile of microglia. Two potential novel microglial PET ligands have been suggested: the ionotropic P2X receptor P2X7, which is expressed in microglia, macrophages and monocytes (Oliveira-Giacomelli et al., 2021), and the metabotropic P2Y receptor P2Y12, which is only expressed by microglia and considered as a homeostatic marker (Beaino et al., 2017; Mildner et al., 2017; Franco-Bocanegra et al., 2019b). Experimental studies have observed P2Y12 expressed by anti-inflammatory microglia and P2X7 by pro-inflammatory microglia. Also, binding of P2X7 PET ligand was increased in multiple sclerosis (MS) active lesions, whereas the signal of the P2Y12-PET ligand was decreased in all MS lesions (active, chronic active and chronic inactive) (Beaino et al., 2017). To our knowledge, the expressions have not been tested in AD. Cannabinoid receptor type 2 (CBR2) has been proposed as another potential target for PET tracers of microglia (Núñez et al., 2004). However, [C11]NE40 (a CBR2 ligand) binding was lower in AD compared to controls and there was no association between the ligand and Aβ plaques (Ahmad et al., 2016), suggesting that this may not be a good target for the identification of activated microglia in disease.
Magnetic resonance imaging has been less utilized than PET to assess gliosis due to its lack of cell specificity. Of note, it was recently suggested that iron associates with microglia in AD, especially in dystrophic microglia defined as senescent or dysfunctional (Streit et al., 2009), and microglia primed to phagocytose Aβ (Kenkhuis et al., 2021). MRI is very sensitive to iron (Zeineh et al., 2015) and therefore might be used to find brain areas with microglial populations involved in AD.
Interestingly, a study using diffusion-weighted MRI built a microstructural model of diffusion based on the ramified morphology of glial cells. The model was tested on rats under several conditions and the protocol was validated for glial cells detection via the immuno-labeling of the brain rats for microglia (Iba1) and astrocytes (GFAP). Interestingly, this novel model appears not only to be able to distinguish between microglia and astrocytes but also to assess the presence of neurodegeneration. The model was then tested on six healthy patients to evaluate reproducibility of the method as a proof-of-concept. Similar patterns of microglial cell density were observed within different regions as those reported in post-mortem tissue (Garcia-Hernandez et al., 2021).
Magnetic resonance imaging has been widely used to assess certain characteristics of AD, such as brain atrophy, but it remains to be perfected for a precise and fully accurate detection of activated glial cells and the development of novel mathematical models are underway.
Astrocyte ImagingAs mentioned above TSPO has been commonly used as a target for PET imaging of activated microglia. However, studies have shown that TSPO is also expressed by other cerebral cells. In AD PET studies, TSPO was increased in macrophages, endothelial cells, vascular smooth muscle cells, and most notably reactive astrocytes (Cosenza-Nashat et al., 2009; Gui et al., 2020). Indeed TSPO appeared to be specifically upregulated in astrocytes and microglia (Pannell et al., 2020), but interestingly, its expression was not related to microglia, astrocytes, Aβ, tau neurofibrillary tangles or cortical thickness in post-mortem AD (Gui et al., 2020). This suggests that a TSPO-PET signal may not solely highlight microglial changes, but also changes in astrocytes.
Astrocyte specific PET tracers have been developed and most commonly target monoamine oxidase B (MAO-B), which is upregulated in activated astrocytes (Ekblom et al., 1993). A common tracer used to detect this protein is 11C-deuterium-L-deprenyl (11C-DED) with the binding increased in AD vs. controls (Santillo et al., 2011), supporting a role for these cells in the disease. This study, however, did not take into account disease severity. When looking at MCI patients with and without amyloid pathology, as well as in AD patients and controls, levels of 11C-DED were increased in cortical and subcortical regions of MCI patients with amyloid pathology compared to control and AD patients (Carter et al., 2012). Testing the 11C-DED tracer on post-mortem tissue also revealed increased binding in brains with tau pathology in Braak Stage I-II and reduced in brains with more severe tau pathology (Gulyás et al., 2011). Findings from both of these studies imply that astrocytes become activated early in the development of AD, but that their activation is lessened as the disease progresses, perhaps due to astrocytic cell pathology. This has also been shown in autosomal dominant AD, where a pre-symptomatic increase in 11C-DED was detected (Rodriguez-Vieitez et al., 2016) along with a gradual decrease in astrocytosis as Aβ deposition (measured by 11C-PiB) rose.
Other common targets developed for the detection of astrocytosis by PET are the type-2 imidazoline receptors (I2Rs). These receptors have previously been shown to be increased in density in the brains of AD patients (Ruiz et al., 1993). The tracer 11C-BU99008 is a highly specific tracer for activated astrocytes in the human brain (Tyacke et al., 2018)
留言 (0)