Cardiogenetics, Vol. 11, Pages 263-289: Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy
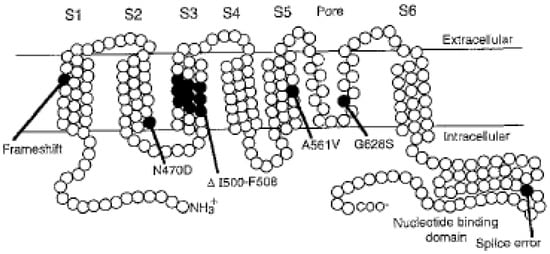
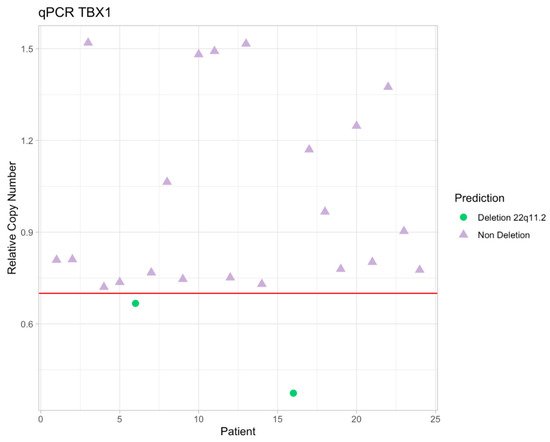
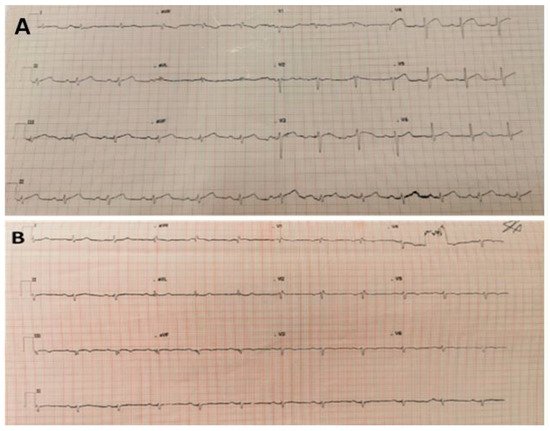
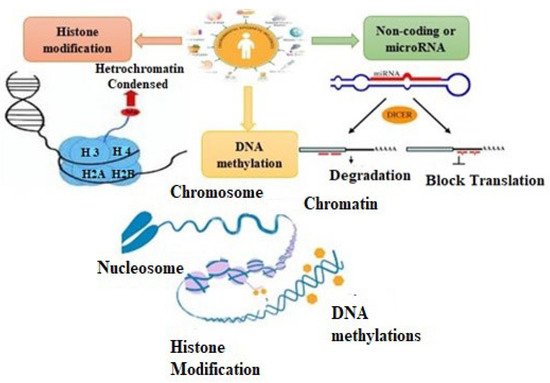
Arrhythmogenic cardiomyopathy (ACM) is a genetically determined myocardial disease associated with sudden cardiac death (SCD). It is most frequently caused by mutations in genes encoding desmosomal proteins. However, there is growing evidence that ACM is not exclusively a desmosome disease but rather appears to be a disease of the connexoma. Fibroadipose replacement of the right ventricle (RV) had long been the hallmark of ACM, although biventricular involvement or predominant involvement of the left ventricle (LD-ACM) is increasingly found, raising the challenge of differential diagnosis with arrhythmogenic dilated cardiomyopathy (a-DCM). A-DCM, ACM, and LD-ACM are increasingly acknowledged as a single nosological entity, the hallmark of which is electrical instability. Our aim was to analyze the complex molecular mechanisms underlying arrhythmogenic cardiomyopathies, outlining the role of inflammation and autoimmunity in disease pathophysiology. Secondly, we present the clinical tools used in the clinical diagnosis of ACM. Focusing on the challenge of defining the risk of sudden death in this clinical setting, we present available risk stratification strategies. Lastly, we summarize the role of genetics and imaging in risk stratification, guiding through the appropriate patient selection for ICD implantation.
View Full-Text
►▼
Show Figures
This is an open access article distributed under the
Creative Commons Attribution License which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited




留言 (0)