Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) Syndrome: Frequency, Clinical Features, Imaging, Histopathologic, and Molecular Genetic Findings in a Third-level Health Care Center in Mexico
Introduction:
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome, is a multisystemic entity of mitochondrial inheritance. To date, there is no epidemiological information on MELAS syndrome in Mexico.
Case Series:
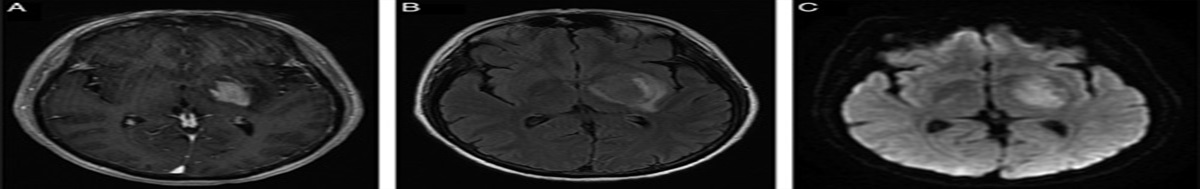
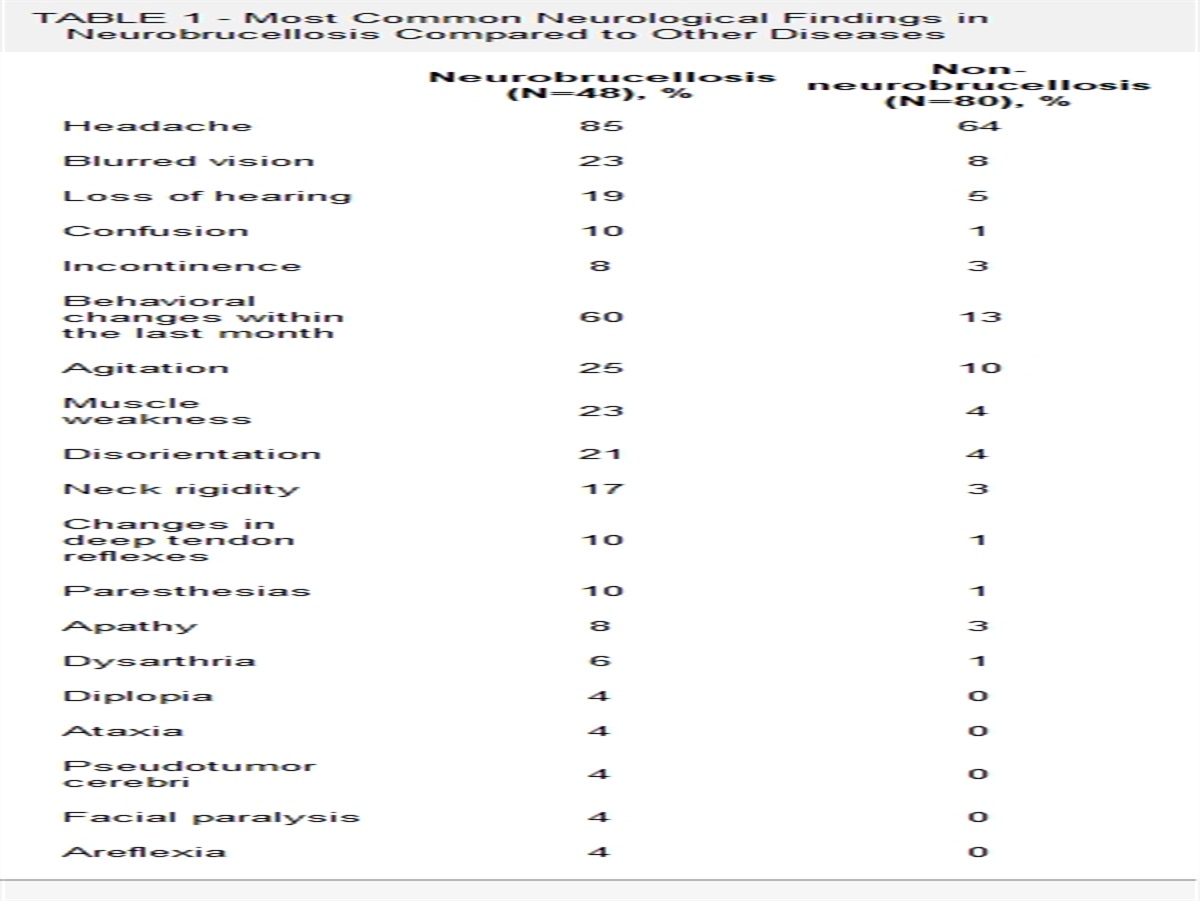
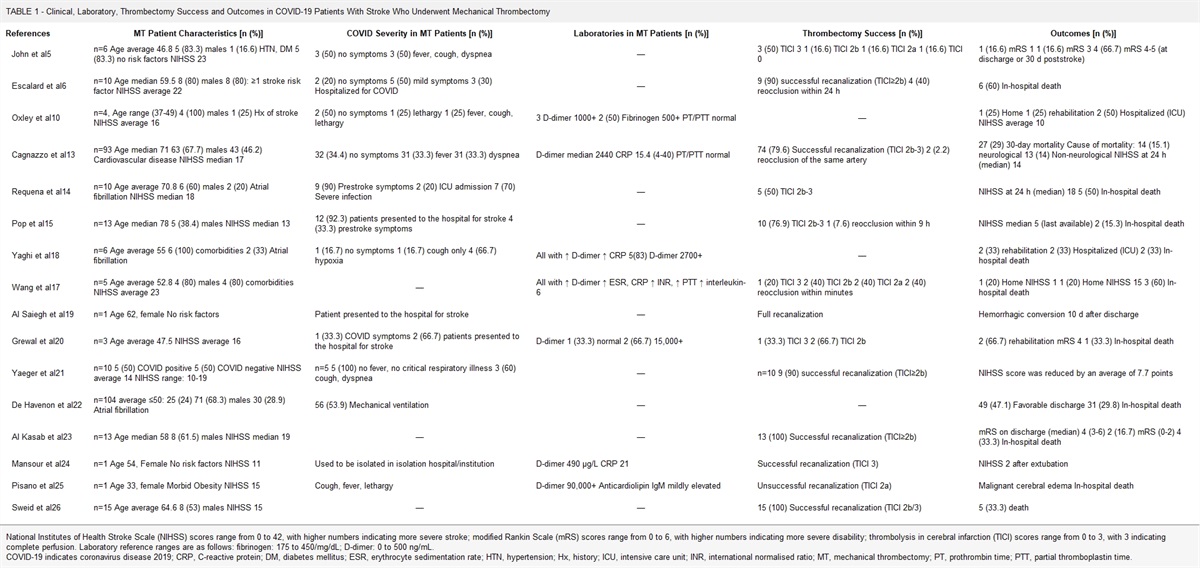
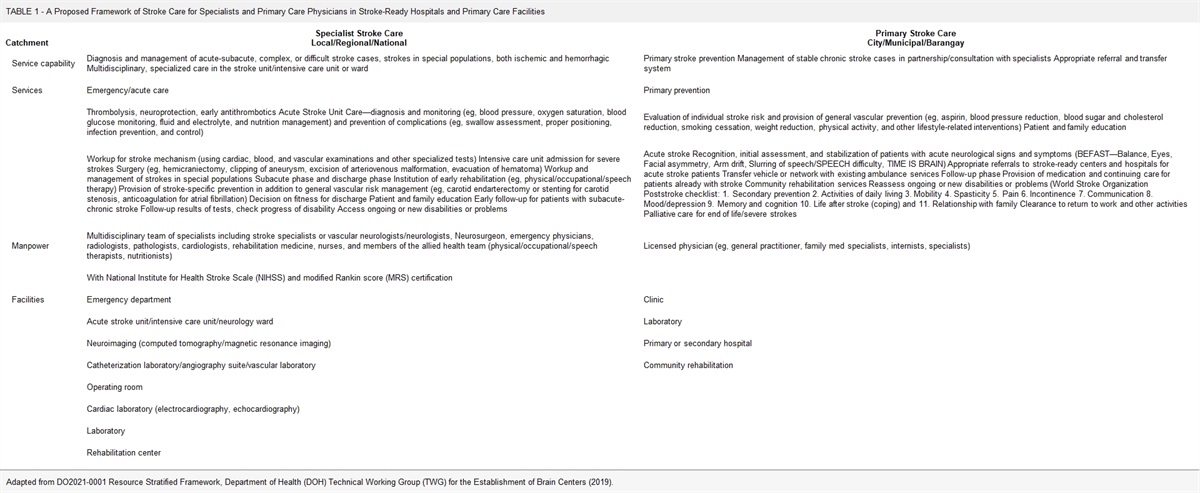
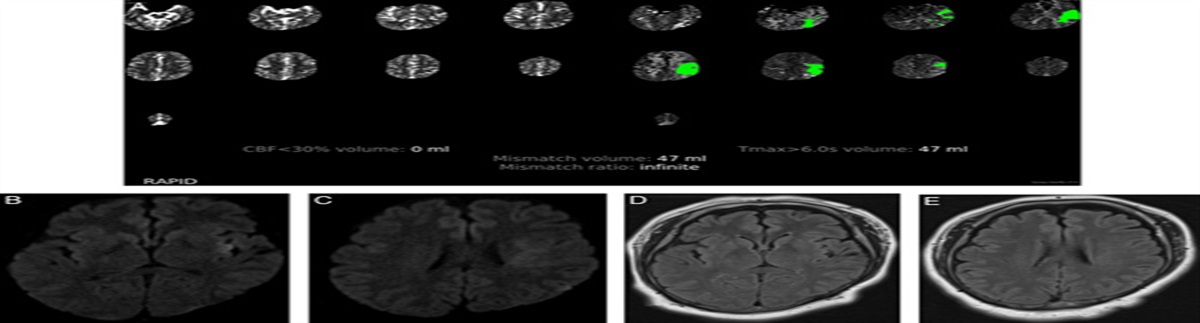
A retrospective, cross-sectional design was employed to collect and analyze the data. The clinical records of patients with mitochondrial cytopathies in the period ranging from January 2018 to March 2020 were reviewed. Patients who met definitive Yatsuga diagnostic criteria for MELAS syndrome were included to describe frequency, clinical, imaging, histopathologic, and molecular studies. Of 56 patients diagnosed with mitochondrial cytopathy, 6 patients met definitive Yatsuga criterion for MELAS (10.7%). The median age at diagnosis was 34 years (30 to 34 y), 2 females and the median time from onset of symptoms at diagnosis 3.5 years (1 to 10 y). The median of the number of stroke-like episodes before the diagnosis was 3 (range, 2 to 3). The main findings in computed tomography were basal ganglia calcifications (33%), whereas in magnetic resonance imaging were a lactate peak in the spectroscopy sequence in 2 patients. Five patients (84%) had red-ragged fibers and phantom fibers in the Cox stain in the muscle biopsy. Four patients (67%) had presence of 3243A>G mutation in the mitochondrial MT-TL1 gene. One patient died because of status epilepticus.
Conclusions:
MELAS syndrome represents a common diagnostic challenge for clinicians, often delaying definitive diagnosis. It should be suspected in young patients with stroke of undetermined etiology associated with other systemic and neurological features.



留言 (0)