
記住我







The patient is an 8-year-old boy born to healthy, non-consanguineous Han Chinese parents following a normal pregnancy and full-term delivery (Fig. 1A). The pregnancy was complicated by turbid amniotic fluid. At birth, he was diagnosed with neonatal pneumonia and epilepsy, receiving 17 days of treatment in the Neonatal Intensive Care Unit (NICU). The patient presented with early global developmental delay, which was characterized by an inability to speak or walk, alongside severe dystonia, choreoathetoid movements, epilepsy, and non-specific facial dysmorphisms such as midface hypoplasia, short philtrum, low-set ears, and thick, wiry hair (Fig. 1C). His older sister also had developmental delays and feeding difficulties. Diagnosed with infantile spasms at around 4 months of age, she unfortunately passed away from renal failure at 6 months. The boy’s younger brother, born in 2023, is currently developing normally. A brain MRI at 8 years old revealed abnormal signals around the lateral ventricles, basal ganglia, and thalamus. It also showed a smaller and thickened corpus callosum, as well as bilateral frontotemporal subarachnoid spaces and deepened sulci, indicating loss of white matter and cerebral hypoplasia (Fig. 1B). Laboratory studies showed mild microcytic anemia, characterized by a hemoglobin level of 114 g/L (normal range: 120–140 g/L) and a mean corpuscular volume of 79.8 fL (normal range: 82–100 fL). Serum ferritin was measured at 24.34 ng/mL (normal range: 21.8–274.66 ng/mL), and iron levels were 7.84 µmol/L (normal range: 7.36–9.34 µmol/L).
Fig. 1
Genetic analysis of IREB2 variants in a Chinese family due to neurodevelopmental delay in the patient. The arrow indicates the patient. A. Pedigree with the IREB2 mutations in this study. B. Representative brain magnetic resonance images (MRI) of the patient. C. Clinical phenotypes and severity observed in the patient. D. Whole-exome sequencing (WES) analysis identified two compound heterozygous missense variants in the IREB2 gene. E. Normal Copy Number Variation Sequencing (CNV-seq) result of the patient. F. Sanger sequencing confirmed the biallelic IREB2 variants in the patient (II:2) inherited from his father (I:1, c.2477 A > T) and mother (I:2, c.1111 A > G). G. PCR products of the Exon regions containing mutation sites and the coding sequence (CDS) of IREB2 gene in this family
Identification and functional analysis of IREB2 mutations in the NDCAMA patientClinical whole-exome sequencing (WES) identified compound heterozygous missense mutations in the NDCAMA-associated IREB2 gene (GenBank: NM_004136.2; c.1111 A > G and c.2477 A > T). These mutations were classified as likely pathogenic according to American College of Medical Genetics and Genomics (ACMG) recommendations (Fig. 1D). Located in exons 9 and 20, these variants have a low population frequency and are not currently included in the HGMD and ClinVar databases. Copy number variant sequencing and inherited metabolic disease screening yielded negative results (Fig. 1E). Sanger sequencing confirmed the paternal (A2477T) and maternal (A1111G) origins of the variants, with PCR amplification of the sequence containing mutated sites and full-length coding sequencing of IREB2 showing no splicing abnormalities (Fig. 1F-G). Both mutations, at positions Ile371 and Asp826, resulted in valine substitutions, which in silico analysis suggests deleterious effects (Fig. 2A). Specifically, P.Ile371Val may impact protein stability, while P.Asp826Val could lead to hydrophobicity alteration and structural change in IRP2 (Fig. 2B). Furthermore, the amino acid sequences at the mutation sites are highly conserved among different mammalian species (Fig. 2C-D). Unfortunately, the patient’s older sister did not undergo genetic testing, while his little brother carried the single A2477T mutation inherited from his father.
Fig. 2
In silico predictions (A-B) and evolutionary conservation (C-D) of P.I371V and P.D826V on IRP2 protein
To further evaluate the impact of these biallelic variants on cellular iron metabolism, we used lentiviral infection to establish wild-type and mutant flag-tagged IRP2-overexpressing SH-SY5Y cells, a human neuroblastoma cell line. High infection efficiency was confirmed through GFP expression (Fig. 3A). Western blot analysis revealed that the A2477T mutation led to an approximate 70% reduction in IRP2 expression, as evidenced by the exogenously transferred flag-IRP2 expression (Fig. 3B). Compared to the wild-type group, the expression of FTH was notably increased while TFRC expression was significantly decreased. Conversely, the A1111G mutation did not impact the expression of iron metabolism proteins, although there was a slight increase in FTH expression (Fig. 3B-C). The intracellular Fe2+ level in the A2477T group was drastically reduced, similar to the control group, while no significant change was observed in the A1111G group. Furthermore, we examined the expression of IREB2 and iron metabolism-related genes in patient-derived PBMCs. The results aligned with the A2477T mutation, showing indistinctive alteration in IREB2 at the mRNA level, thus ruling out IREB2 splicing abnormalities proposed in previously reported patients (Fig. 3E). The degradation of IRP2 protein plays an important role in regulating intracellular iron metabolism homeostasis [23]. Treatment with the proteasome inhibitor MG-132 led to IRP2 restoration and iron metabolism-related proteins’ expression, resulting in a remarkable increase in Fe2+ concentration (Fig. 3F-G). The findings suggest that the A1111G mutation is relatively mild, while the degradation of the IRP2 protein due to the A2477T mutation may play a key role in the pathogenesis of NDCAMA in this patient.
Fig. 3
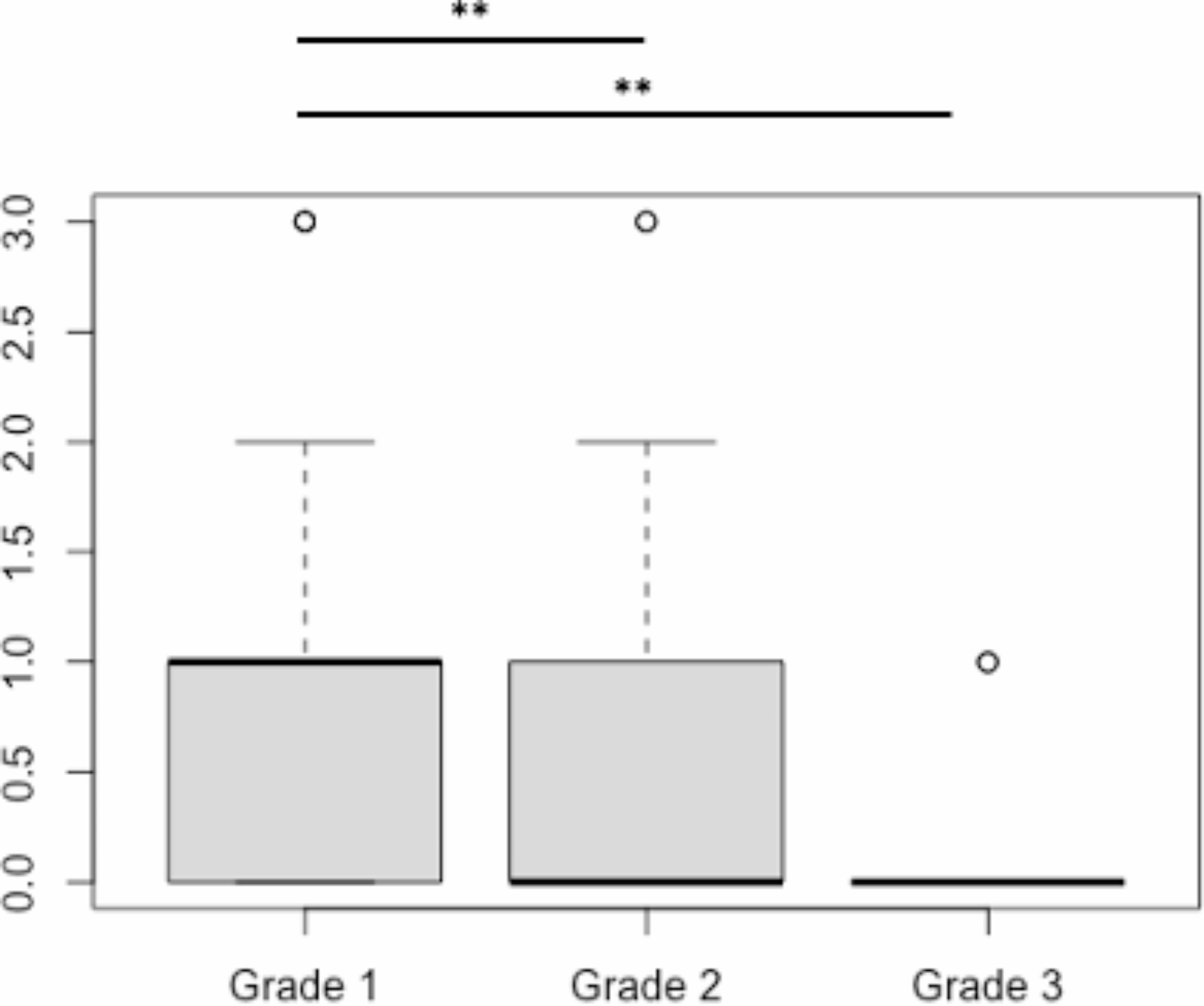
The impact of biallelic IREB2 mutations on iron metabolism. A. SH-SY5Y cells were infected with lentivirus carrying Flag-tagged IREB2-wide-type (WT), A1111G, A2477T or empty vector (Blank), Scare bar = 100 μm. B. Expression of IREB2-regulated iron metabolism genes were detected by Western blot. C. Quantification of the Western blot results (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001). D. Intracellular iron in different group was detected by the utilization of a FerroOrange fluorescent probe. E. RT-qPCR was performed to quantify mRNA levels of iron metabolism genes in healthy individual and the patient (n = 5, ***p < 0.001). F. Quantification of the western blot results to detect the protein levels of IRP2 and iron metabolism-associated proteins in A2477T cells treated with 40µM MG-132 or DMSO (n = 3, **p < 0.01, ***p < 0.001). G. Fluorescence intensity of intracellular iron levels in MG-132 treated cells or NC group (n = 5, **p < 0.01)
Summary of reported cases and functional analysis of IREB2 mutations in NDCAMAA literature review has identified four reported cases of IREB2-associated NDCAMA (Fig. 4A, Table S2). Costain et al. (2019) reported the first case of a 16-year-old boy born to unrelated Filipino parents [18]. The second case, described by Cooper et al. (2019), involved a 10-year-old boy born to unrelated Australian parents [19]. Lastly, Maio et al. (2022) presented the third case of a 7-year-old boy born to healthy, non-consanguineous parents of Sephardic and Sephardic/Irish descent in the USA [20]. All three previously reported cases, along with our patient, exhibited similar clinical features, including neonatal feeding difficulties, hypotonia, choreoathetoid movements, impaired ambulation and communication, and non-specific facial dysmorphisms. Furthermore, brain imaging studies revealed progressive cerebral volume loss, delayed myelination, and a reduction in white matter volume. Notably, electroencephalograms (EEG) were abnormal in all three patients, although clinical seizures were not observed in the second patient described by Cooper et al. Additionally, laboratory studies indicated mild microcytic anemia, while serum iron levels remained within the normal range.
Fig. 4
Known pathogenic variants in patients diagnosed with NDCAMA. A. Detailed view of the distribution of pathogenic IREB2 variants (GenBank: NM_004136.4) in 4 reported cases with base changes in purple and amino acid changes in red. Two representative views of the crystal structure including IRP2 binding to iron-responsive element (IRE) of ferritin (B), or FBXL5 (C) based on 6VCD and 3SNP. The amino acid residues mutated were depicted in red
Whole-exome sequencing, or parallel gene sequencing, revealed that the patients were compound heterozygous for two variants in IREB2. The first case exhibited two nonsense mutations (c.1069G > T/p.Gly357* and c.1255 C > T/p.Arg419*), while the second case presented a missense mutation (c.2353G > A/p.Gly785Arg) accompanied by a 3-base pair in-frame deletion (c.1329_1331del/p.Ser444del). The third case, along with our patient, displayed two missense mutations (c.656 A > C/p.Glu219Ala and c.2240G > A/p.Gly747Glu). Cellular studies utilizing patient-derived lymphoblasts demonstrated a complete loss of IRP2 expression in the first case, resulting in altered post-transcriptional regulation of iron metabolism genes including FTH and TFRC, accompanied by a significant reduction in iron levels, which was primarily attributed to the mRNA surveillance pathway known as nonsense-mediated mRNA decay (NMD). Although cellular studies were not performed for the second case, in silico analyses indicated the potentially deleterious effects of the two mutations on iron-responsive element (IRE)-binding activity. In contrast, a dramatic decrease in IRP2 protein levels was observed in the patient-derived cells from the third case, where the patient also exhibited severe clinical symptoms. In the discussion, the authors proposed that mis-splicing of IREB2 mRNA and altered IRE-binding activities may elucidate the pathogenesis observed in the third patient. While these three cases were investigated directly using patient-derived cells, there remains a notable absence of experimental evidence to assess the detrimental effects of each mutation.
Unlike previous studies that examined compound heterozygous mutations, this study investigated the functions of the two mutations separately and identified c.2477 A > T mutation as deleterious, resulting in the degradation of IRP2. In contrast, the c.1111 A > G mutation appears to primarily affect iron-responsive element (IRE)-binding activity, which we classify as a relatively mild mutation.
Through modular docking and structure-based prediction, we identified the distribution pattern of compound heterozygous variants in NDCAMA patients. One mild mutation was located close to functional domains where IRP2 binds to the IRE element of iron metabolism-related mRNA, while the severe one was near the region where IRP2 binds to FBXL5, a key protein involved in IRP2 degradation (Fig. 4B). These findings may explain how compound heterozygous missense mutations contribute to abnormal iron metabolism in DNCAMA patients.
留言 (0)