
記住我







A total of 120 messenger RNAs (mRNAs) related to the interaction between sialic acid and siglec were initially acquired from the MsigDB website. A total of 2511 potential lncRNAs associated with sialylation were found using the Pearson correlation test, employing a threshold of |r| > 0.3 and P < 0.001.
In order to assess the prognostic relevance of the potential lncRNAs, we utilized 413 lncRNAs from TCGA with complete clinical data. Following a random allocation of the 413 patients into distinct training and validation groups, we conducted univariate Cox regression analysis, with a p-value threshold of 0.001. The present investigation resulted in the identification of 14 lncRNAs that are related with sialylation and exhibit prognostic importance. The further screening of these variables was conducted using lasso regression in order to proceed with the subsequent stage, which entailed multifactorial Cox regression analysis. (Fig. 1A, B).
Fig. 1
Identifying 6 sialylation-related Long noncoding RNAs (lncRNAs)s. (A) Profiles of Lasso coefficients. (B) Cross-validation for tuning parameter selection
Subsequently, a comprehensive investigation was conducted to identify and construct a model for a set of six lncRNAs that are connected with sialylation. The prognostic significance of these lncRNAs was determined using multifactorial Cox regression analysis. Several genes were identified as negative prognostic factors, including LINC01508, AC012625.1, AL135905.1, GRASLND, and ARHGAP5-AS1. Conversely, AC104785.1 was determined to be a favorable prognosis factor (Fig. 2A).
These 6 sialylation-related LncRNAs were used to create a TIL-related LncRNA signature with the following risk score formula: Risk score = (0.097657 × expression level of LINC01508) + (-0.3505 × expression level of AC104785.1) + (0.230 × expression level of AC012625.1) + (0.2500 × expression level of AL135905.1) + (0.2936 × expression level of GRASLND) + (0.1070 × expression level of ARHGAP5-AS1).
Subsequently, we evaluated the predicted survival values of the model by plotting ROC curves for 1, 3, and 5 years. The area under the curve (AUC) values were 0.693, 0.698, and 0.688 (Fig. 2B). The AUC values for the risk model are not sufficiently high at 1, 3, and 5 years. This could be due to factors such as sample size, feature variables, and the chosen machine learning algorithms. Notably, the AUC values of the 1-year curves were significantly higher than other clinical parameters, demonstrating that the model developed using the risk score reliably predicted survival (Fig. 2C).
Fig. 2
Recognition of Prognostic sialylation-related lncRNAs and Model Development. (A) Multivariate Cox model of 6 sialylation-related lncRNAs. (B) ROC curves for 1, 3, and 5 years. (C) The 1-year ROC curves of the risk score and other clinical characteristics
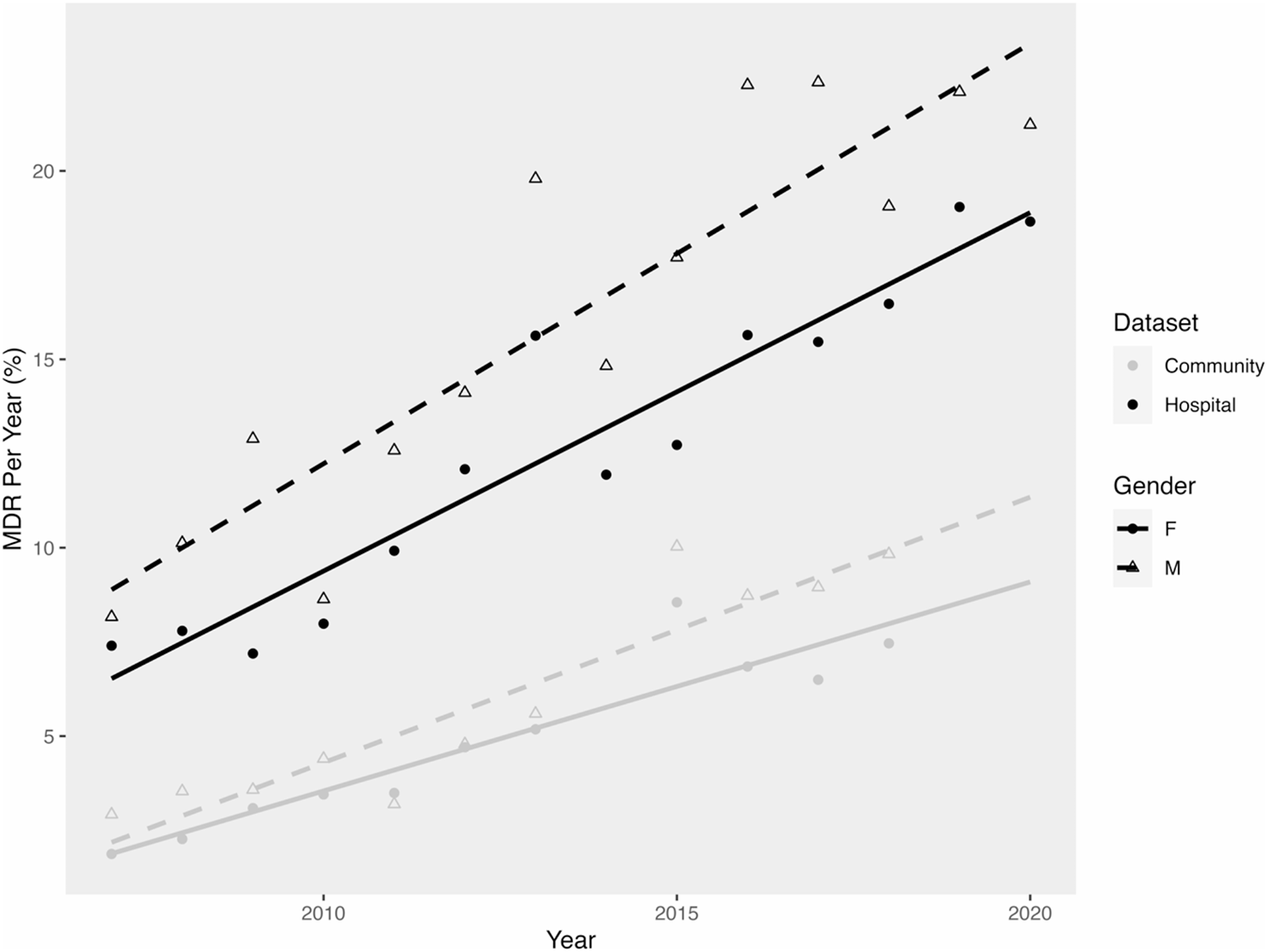
Value assessment of prognostic sialylation-related LncRNAsPatients were categorized as either high-risk or low-risk based on the median of the calculated risk scores in each group. In both the training group (Fig. 3, left), the test group (Fig. 3, middle), and the combined group (Fig. 3, right), the high-risk group exhibited higher mortality rates, indicating a less favorable prognosis for patients with high-risk scores (p < 0.001). The risk scores and survival status of the training, test, and combination groups are displayed in Fig. 3(bottom).
The results shown in Fig. 4A highlight a significant difference between stage II and stage III (p = 0.00094) and between stage II and stage IV (p = 0.00037). Furthermore, Fig. 4B reveals that risk scores were lower in stage N0 compared to stage N2 (p = 0.028). Additionally, Fig. 4C demonstrates that T2 had significantly lower scores than T3 (p = 0.0018) and also lower than T4 (p = 0.019). Additionally, the prognostic value of the risk score was evaluated in patients from different stratified cohorts based on age, gender, and tumor stage. For this evaluation, we only included subgroups with 50 or more patients after stratification. The results indicated that the risk score effectively predicted prognosis in various cohorts, including age ≤ 65, age > 65, female, male, stage I/II, and stage III/IV (p < 0.001)(Fig. 4D).
To explore the relationship between clinicopathologic characteristics and risk scores, we utilized the chi-squared (c2) test and Cox regression in the combined group. The c2 test indicated differences in grade, clinical stage, and T stage between the high-risk and low-risk groups (Fig. 5A). Univariate Cox regression analysis revealed significant associations of stage (HR 1.643, 95% CI: 1.255–2.152, p < 0.001) and risk score (HR 1.084, 95% CI: 1.055–1.114, p < 0.001) with the outcome (Fig. 5B). Furthermore, multivariate Cox regression analysis confirmed a similar association between stage (HR 1.637, 95% CI: 1.241–2.160, p < 0.001) and risk score (HR 1.087, 95% CI: 1.059–1.116, p < 0.001) and the outcome, underscoring the significant connection between risk scores and overall survival (Fig. 5C).
Fig. 3
Distribution of risk score (high or low) and status (dead or alive) and KM curves of OS in the training (left), testing (medium), and combined (right) sets
Fig. 4
Significant differences between between risk scores and clinical characteristics in TCGA-BLCA cohort. Differences between risk score and tumor stage (A), T stage (B), N stage (C). (D) Survival analysis of the sialylation-related lncRNA signature in different stratified cohorts
Fig. 5
Value assessment of prognostic sialylation-related lncRNAs. (A) The result of chi-squared test. (B) Univariate Cox regression analysis. (C) Multivariate Cox regression analysis
Potential mechanism analysis of the sialylation-related LncRNA signatureWe conducted KEGG pathway, GSEA, and GO analyses to explore the underlying mechanisms by which the risk signature stratifies patient prognosis. Initially, we employed pathway analysis and GSEA to uncover the biological significance of the sialylation-related lncRNA signature. Through the use of the R package “edgeR,” we identified a total of 612 DEGs with |log2(FC)| > 1 and adjusted P < 0.05 between the two risk groups in the combined data.
According to the GSEA analysis, the high-risk group exhibited enrichment in the top five pathways. These pathways were identified as cytokine-cytokine receptor interaction, extracellular matrix (ECM)-receptor interaction, focal adhesion, neuroactive ligand-receptor interaction, and regulation of actin cytoskeleton (Fig. 6A). Conversely, the top five enriched pathways in the low-risk group encompassed alpha-linolenic acid metabolism, drug metabolism - cytochrome P450, linoleic acid metabolism, primary immunodeficiency, and ribosome (Fig. 6B). In addition, the GO enrichment analysis conducted on the 612 DEGs indicated their participation in many biological processes, including epidermis formation, epidermal cell differentiation, and granulocyte chemotaxis (Fig. 6C).
Fig. 6
Potential mechanism analysis of the sialylation-related LncRNA signature. (A) The top five enriched pathways in the low-risk group. (B) The top five enriched pathways in the low-risk group. (C) The result of the GO enrichment analysis
Construction and validation of a sialylation-related LncRNA prognostic modelThe column line graphs present data on risk score, age, lymph node metastatic stage, and pathological grading. The column line plot demonstrates a clear association between the risk score and OS in patients with BLCA (Fig. 7A).
The nomogram’s C-index was found to be 0.693, indicating the predictive accuracy of the model. Additionally, the calibration curves exhibited a high level of concordance between the projected probability and the observed outcomes. It is worth mentioning that the calibration curves for the OS at 1-year, 3-year, and 5-year intervals (Fig. 7E) exhibited a tight adherence to the 45-degree line. In addition, the nomograms exhibited area under the curve (AUC) values of 0.778, 0.743, and 0.748 for the 1-year, 3-year, and 5-year OS periods, as illustrated in Fig. 7B-D. The nomograms exhibit superior performance compared to individual clinical predictors, as indicated by their high AUC values. This suggests that the integration of many risk indicators can improve the prognostic accuracy for BLCA. PCA revealed the distribution of the two risk groups along two axes. This observation suggests that the riskscore-associated lncRNAs exhibit a more effective classification of BLCA patients into high-risk and low-risk groups (Fig. 7F), compared to sialylation-related lncRNAs (Fig. 7G), sialylation -related genes (Fig. 7H), and all genes (Fig. 7I). These findings highlight the superior discriminative ability of the risksocre-associated lncRNAs in the identification process.
Fig. 7
Construction and validation of a sialylation-related LncRNA prognostic model. (A) Nomogram constructed by independent prognostic factors. (B-D) ROC curves show the predictive accuracy of the nomogram, risk score, and other clinical characteristics. (E) Calibration curves for the OS at 1-year, 3-year, and 5-year intervals
(F) Principal component analysis (PCA) of the riskscore-related lncRNAs. (G) PCA of the sialylation-related lncRNAs. (H) PCA of the sialylation -related genes. (I) PCA of the all genes.
Assessing the immune microenvironment of sialylation-related signature scoreIn order to delve deeper into the relationship between the immune process and risk scores, we conducted an assessment of lymphocyte lines using CIBERSORT. The results of the CIBERSORT analysis (Fig. 8A), indicate that the high-risk group demonstrated a greater prevalence of activated T cells CD4 + memory resting Macrophages M0. Conversely, the low-risk group exhibited a larger prevalence of active Plasma cells, CD8 + T cells, and regulatory T cells (Tregs). The correlation between lymphocyte populations and sialylation-related lncRNAs is depicted in Fig. 9A. Significantly, there were high associations observed between AC104785.1 and Treg, T cells CD4 + memory activated, Plasma cells, and Macrophages M1. Additionally, a robust link was found between AC012625.1 and Plasma cells. Furthermore, there was a significant association observed between T cells follicular helper and Macrophage M1 with GRASLND. Similarly, T cells CD4 memory resting, T cells CD4 memory activated, and mast cells resting exhibited a high correlation with ARHGAP5-AS1.
Our analysis revealed significant variations in immune function between the high and low-risk groups, particularly for APC_co_inhibition, Macrophages, MHC_class_I, Parainflammation, TIL, and Treg, as demonstrated in Fig. 8B. In addition, our investigation involved the evaluation of a total of 79 genes related with immune checkpoint. Among these, 47 genes displayed differences between the high and low-risk groups, as depicted in Fig.S1. Furthermore, the analysis of Fig. 9B demonstrates a greater occurrence of C1 subtypes in the low-risk groups, while the high-risk groups exhibit a higher proportion of C2 subtypes (P = 0.001).
Fig. 8
Immune cell infiltration and immune function of the different risk groups. (A) Immune cell infiltration of the high and low-groups. (B) Immune function of the high and low-risk groups
Fig. 9
Immunoscape of sialylation-related lncRNAs and risk grops. (A) The correlation between lymphocyte populations and sialylation-related lncRNAs. (B) Immune subtypes of the high and low-risk groups
Tumor mutational burden and drug sensitivity analysisBoth in the high-risk (HR) and low-risk (LR) group, TP53, TTN, KMT2D, MUC16, ARID1A and KDM6A exhibited mutation rates exceeding 20% (Fig. 10A, B). Patients with low tumor mutation burden (TMB) had shorter OS compared to those with high TMB (Fig. 10C). Notably, when considering both risk score and TMB, it was evident that patients with low TMB and high risk exhibited the poorest prognosis (Fig. 10D).
In order to conduct a thorough evaluation of therapeutic efficacy in response to various chemotherapeutic drugs, we calculated the IC50. Our results showed that Sorafenib was less sensitive in the high-risk group, while it was more sensitive in the low-risk group compared to Cisplatin, Docetaxel, and Dasatinib. Therefore, our risk profiles can serve as a valuable tool in evaluating chemotherapeutic drug sensitivity (Fig. 10E-H).
Fig. 10
Tumour mutational burden (TMB) and Drug susceptibility analysis. (A) The waterfall chart of the frequently mutated genes in the high-risk group. (B) The waterfall chart of the frequently mutated genes in the low-risk group. (C) Kaplan–Meier curves of high- and low-TMB on OS. (D) Kaplan–Meier curves of TMB and risk score on OS. exploration of the risk signature and drug sensitivity. (E) Docetaxel. (F) Sorafenib. (G) Dasatinib. (H) Cisplatin
Expression validation of sialylation-related LncRNAs signatureThe expression of model-associated lncRNAs was confirmed at the cellular level through quantitative real-time polymerase chain reaction (qRT-PCR). In bladder cancer (BCa) cell lines, including ARHGAP5-AS1, GRASLND, and LINC01508, their expression levels were notably elevated when compared to human urinary epithelial SV-HUC-1 cell lines (Fig. 11A-C).
Fig. 11
Relative expression of lncRNAs. (A) ARHGAP5-AS1. (B) GRASLND. (C) LINC01508
留言 (0)