Study Design and Patients
This was a multicenter, open-label, phase II study that recruited patients from 12 academic medical centers in Japan. Inclusion criteria were age 20–75 years, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, adequate organ function (i.e., absolute neutrophil count ≥1500 cells/μL, white blood cell count ≤12,000 cells/μL, platelet count ≥75,000 cells/μL, total bilirubin ≤2.0 mg/dL, aspartate aminotransferase and alanine aminotransferase ≤100 IU/L, serum creatinine ≤1.2 mg/dL, and creatinine clearance ≥50 mL/min), and provision of written informed consent. Patients had clinical stage IB–III (excluding T4) disease based on the 7th Union for International Cancer Control (UICC) TNM classification of histologically confirmed ESCC, adenosquamous cell carcinoma, or basal cell carcinoma. Patients received neoadjuvant chemotherapy consisting of 5-fluorouracil plus cisplatin and were expected to undergo R0 resection by esophagectomy. All patients registered before surgery. The study protocol was approved by the Ethics Committee and the Institutional Review Board of each institution and was conducted in accordance with the ethical principles originating from the Declaration of Helsinki. This trial was registered at the Japan Registry of Clinical Trials with the number jRCTs051180154 (https://jrct.niph.go.jp/latest-detail/jRCTs051180154).
Procedures
Total or subtotal thoracic esophagectomy, or thoracoscopic esophagectomy, and regional lymphadenectomy were performed after completion of neoadjuvant chemotherapy. Adjuvant chemotherapy was performed within 56 days of surgery. Patients received adjuvant therapy consisting of oral S-1 twice daily for 4 weeks, followed by a 2-week rest period. Three dose levels of S-1 were administered according to body surface area (BSA): <1.25 m2, 40 mg twice daily; 1.25 to <1.50 m2, 50 mg twice daily; and ≥1.5 m2, 60 mg twice daily. Patients with creatinine clearance levels of 50–60 mL/min received lower S-1 doses (BSA <1.25 m2, 25 mg twice daily; 1.25 to <1.50 m2, 40 mg twice daily; and ≥1.5 m2, 50 mg twice daily). Treatment was continued for up to four 24-week cycles. To start each cycle of S-1, patients had to satisfy the following criteria: absolute neutrophil count ≥1200 cells/μL, platelet count ≥75,000 cells/μL, total bilirubin ≤3.0 mg/dL, aspartate aminotransferase and alanine aminotransferase ≤150 IU/L, serum creatinine ≤1.2 mg/dL, and no other non-hematological adverse events grade ≥1 for which an investigator judged administration to be inappropriate.
In addition to the criteria for stopping and restarting S-1 in each cycle, further administration of S-1 in the ongoing cycle was suspended if any of the following adverse events were observed: absolute neutrophil count <1000 cells/μL; platelet count <70,000 cells/μL; grade ≥2 gastrointestinal disorders such as diarrhea, nausea, vomiting, anorexia, or oral mucositis; and no other grade ≥3 non-hematological adverse events. Once administration of S-1 was suspended, the daily dose of S-1 for the next cycle was reduced from 120 mg to 100 mg, from 100 mg to 80 mg, or from 80 mg to 50 mg once daily, depending on BSA, or the administration period was changed from 4 weeks of each 6-week cycle to 2 weeks of each 3-week cycle. All patients were evaluated by computed tomography every 6 months until recurrence or withdrawal of consent. Adverse events were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0.
Statistical Analysis
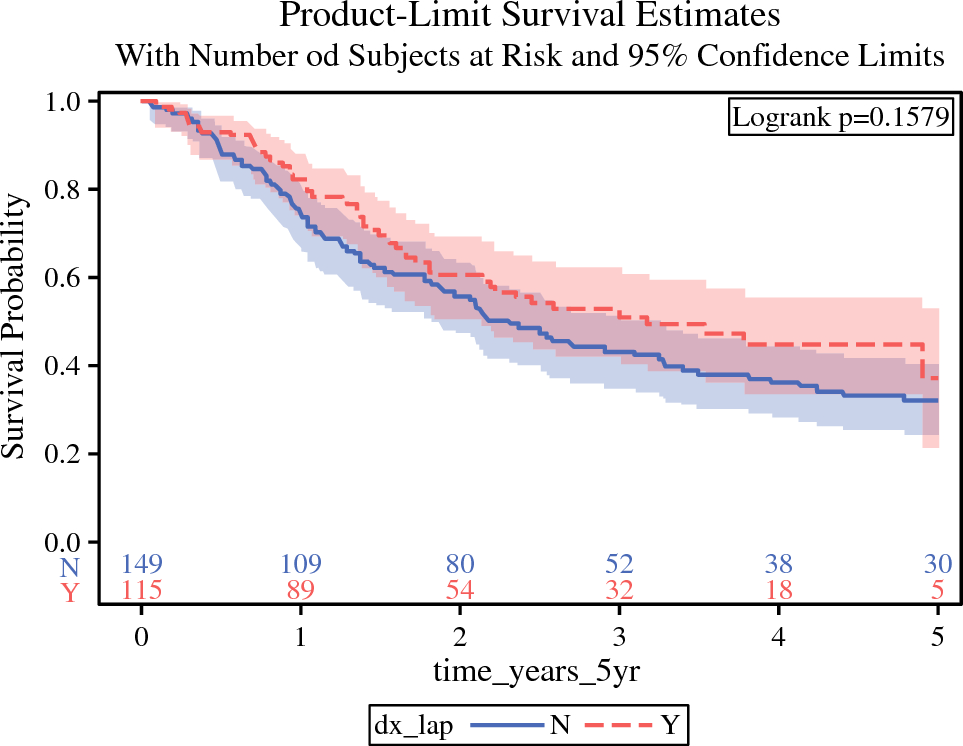
All survival analyses were conducted for all eligible patients and all R0-resected patients. Treatment delivery and safety were assessed in all treated patients. The primary endpoint was 3-year RFS rate, and secondary endpoints were OS, RFS, treatment completion rate, treatment continuation rate per time point, incidence of adverse events, and incidence of treatment-related deaths. OS was defined from the date of esophagectomy to the date of death due to any cause, censored as of the last date the patient was documented to be alive. RFS was defined from the date of esophagectomy until relapse or death from other causes, censored as of the last date the patient was documented to be alive without any evidence of relapse. Incomplete resection was not regarded as an event or censoring due to no relapse. Time-to-event distributions were estimated using the Kaplan–Meier method, and CIs were calculated using Greenwood’s formula. For comparisons of patient subgroups, a univariate Cox proportional hazards model was used. To explore prognostic factors in subgroups, analyses were performed according to sex, age (<65 years vs. ≥65 years), performance status (0 vs. 1), surgical method (open transthoracic esophagectomy vs. thoracoscopic esophagectomy), cT (cT1-2 vs. cT3), cN (cN0 vs. cN1-3), and tumor location (middle thoracic vs. upper thoracic vs. lower thoracic). Statistical analysis was performed using STATA version 17 (StataCorp LLP, College Station, TX, USA).
A minimum sample size of 43 patients with R0 resection was required to provide a power of 0.80 with a one-sided significance level of 0.10, and to detect an alternative 3-year RFS rate of 66% compared with a null hypothesis of 50% on the binomial distribution, according to calculations using PASS software (PASS 11; NCSS, Kaysville, UT, USA). The 3-year PFS rate from the date of surgery in 152 patients who received at least one course of neoadjuvant chemotherapy followed by surgery was 51.2% based on available data from JCOG9907 (unpublished data), therefore we set the threshold at 50%. A total of 50 patients who were expected to undergo R0 resection by esophagectomy was planned for enrolment, with some withdrawals due to R1–2 resection.



留言 (0)