
記住我






This study was a prospective, open-label, multicenter, randomized clinical trial with a 16-week follow-up. Participants were recruited at 15 secondary or tertiary hospitals in China (Beijing Hospital; Yinchuan First People's Hospital; Beijing Aerospace General Hospital; Peking University Shougang Hospital; Haidian Hospital of Beijing, Beijing Boai Hospital; Emergency General Hospital; Zhejiang Provincial People's Hospital; The First Affiliated Hospital of Zhejiang University School of Medicine, Beijing Hepingli Hospital, General Hospital of Civil Aviation, Anyang District Hospital of Puyang City, Zhumadian Central Hospital, Hebi Coal General Hospital, and The First People's Hospital of Qujing City). The study protocol was reviewed and approved by the local ethics committee. All procedures followed the Helsinki Declaration of 1964, as revised in 2013. Comprehensive information is provided to female participants of reproductive age during the screening phase. Participants who express reluctance to adhere to contraceptive requirements are excluded from the study. Throughout the follow-up period, clinical doctors advise female participants of reproductive age and assess their willingness to continue participating in the study. Informed, written consent was obtained from all participants in the study. This trial is registered with the Chinese Clinical Trial Registry (ChiCTR1900028606).
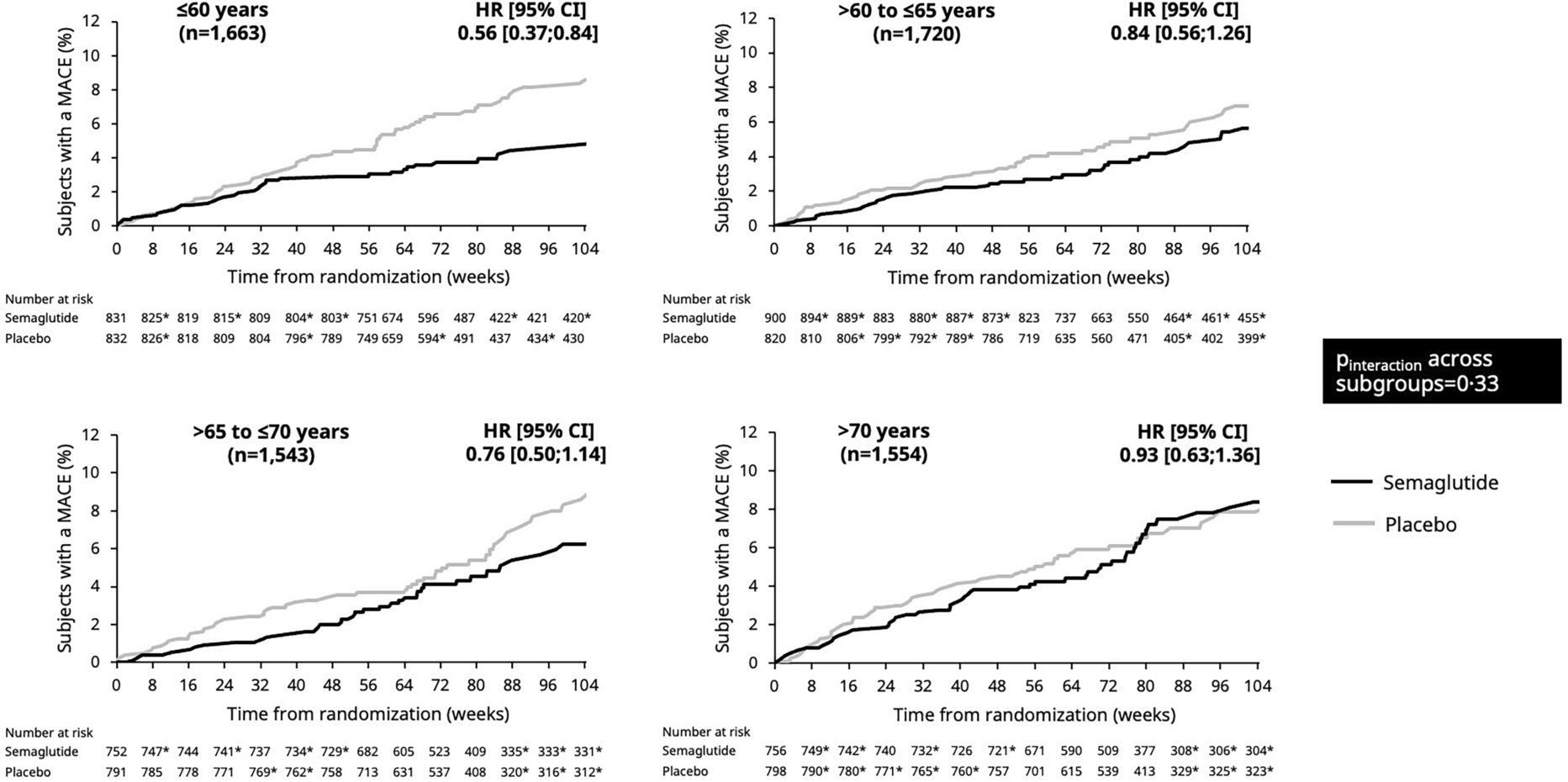
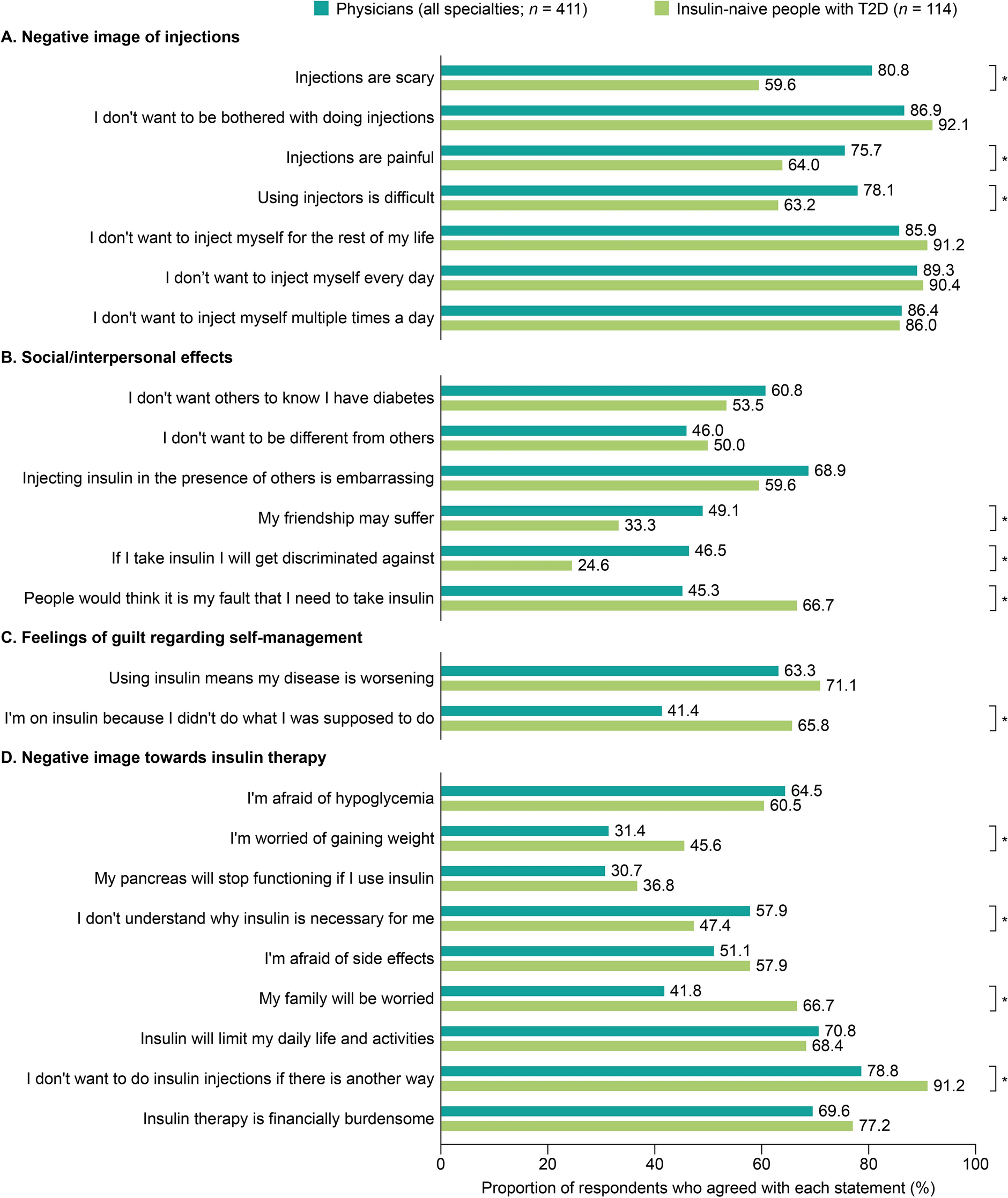
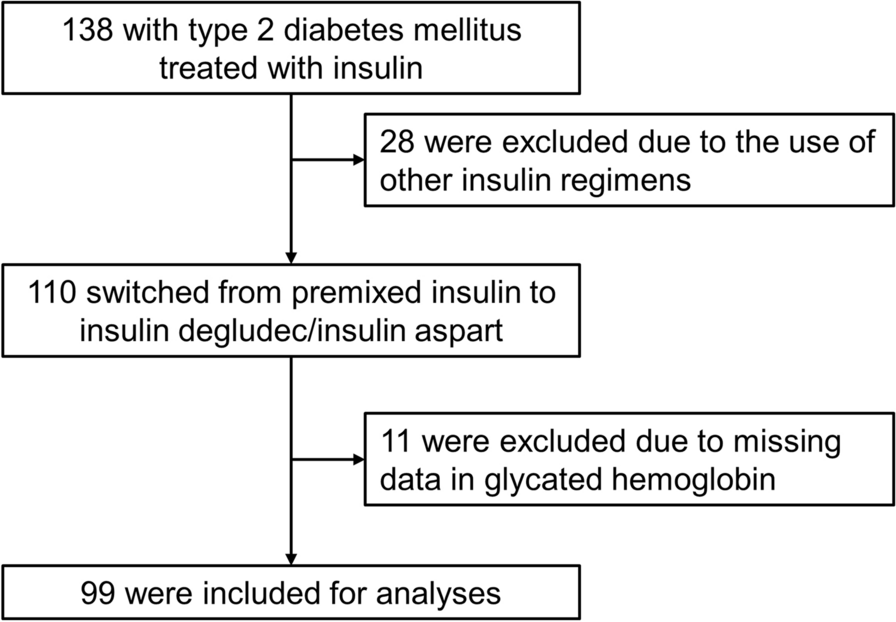
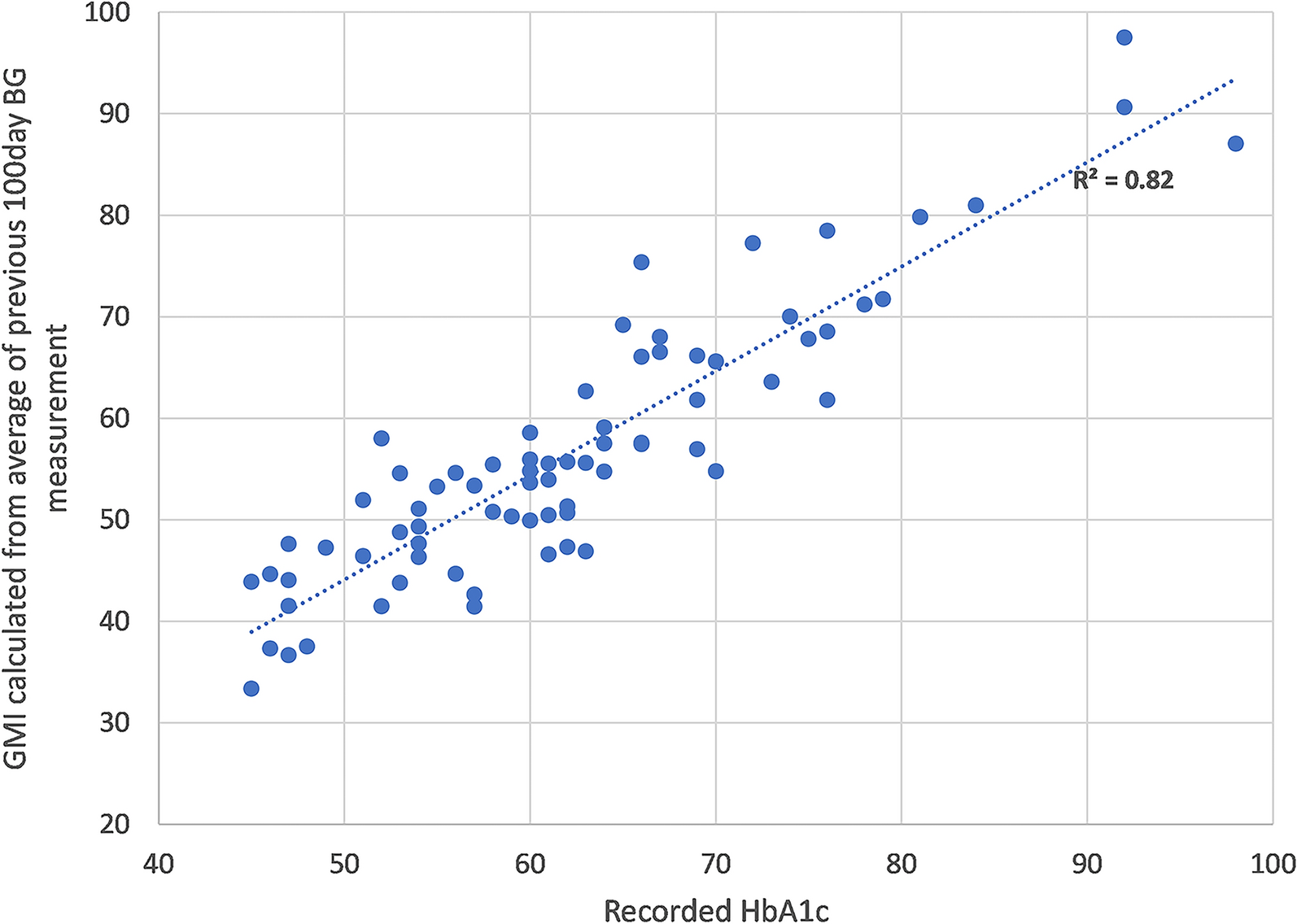
ParticipantsBetween May 6, 2020 and March 1, 2022, 304 of 335 participants were enrolled in this trial. Inclusion criteria encompassed the following: (1) individuals with T2DM aged between 18 and 65 years; (2) body mass index (BMI) ranging from 19 to 45 kg/m2; (3) undergoing monotherapy with metformin of 750–1500 mg with poor glycemic control (7.5 ≤ glycated hemoglobin A1c [HbA1c] ≤ 10%, or fasting blood glucose [FBG] of ≥ 7 and ≤ 13.9 mmol/l). Exclusion criteria included: (1) intolerance of a daily 1500 mg of metformin; (2) a history of hypertension: Systolic blood pressure (SBP) > 180 mmHg and/or Diastolic blood pressure (DBP) > 110 mmHg; (3) a history of severe cardiovascular disease, renal insufficiency, pregnancy, chronic pulmonary disease, or chronic gastrointestinal disease; (4) pregnant women. The flow of screening and recruitment of study subjects is presented in Fig. 1. Patients were 1:1 assigned to two groups stratified in blocks by the baseline HbA1c (< or ≥ 8.5%). Randomization was performed centrally with an interactive web response system. All patients were fully aware of their physical condition, disease diagnosis, treatment protocol, and potential risks and complications throughout the study.
Fig. 1
Flowchart of randomization and enrollment. Poor compliance refers to a participant's failure to consistently adhere to the prescribed investigational drug regimen as indicated by a compliance rate falling below 80% or exceeding 120%. Compliance is calculated by comparing the actual amount of the drug taken by the participant to the prescribed drug amount, expressed as a percentage
Treatment ProtocolParticipants in the control group received a 16-week treatment of 2500 mg/day metformin (Merck & Co Inc), or a maximum-tolerated dose (2000–2500 mg/day) with gradual increments within 0–2 weeks. Participants in the test group received a 16-week treatment of 15/500 mg pioglitazone/metformin (Hangzhou Zhongmei Huadong Pharma Ceutical Co., Ltd) with meals three times daily (twice a day if not well tolerated). During the screening visit, all concomitant medications (baseline recording) were recorded in detail. An explicit record of antidiabetic medication was maintained from the beginning of the randomization until the end of the study. Medications that affect glucose metabolism were not allowed to be used during the observation period, except for the investigational drugs Patients not adhering to combination therapy were excluded from the PPS group. To check for medications that needed to be continued for comorbidities and concomitant medication, doctors requested that the subjects bring all medications they were taking to the follow-up visits. Any other treatments were recorded throughout the study, including the name (or other therapy name), dosage, frequency of use, and time of use.
Study Endpoints and Outcome MeasurementsThe primary endpoint of the current study was the proportions of patients with HbA1c ≤ 6.5% or ≤ 7% at week 16. The secondary endpoints included bodyweight, BMI, FBG, HbA1c, fasting insulin (FI), 2-h postprandial blood glucose (2 h PPBG), fasting capillary blood glucose (FCBG), homeostatic model assessment of insulin resistance (HOMA-IR), SBP and DBP, total cholesterol (TC), triglycerides (TG), free fatty acids (FFA), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), alanine aminotransferase (ALT), aspartate aminotransferase (AST), C-reactive protein (CRP), high-sensitivity C-reactive protein (Hs-CRP), and tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) at week 16. Adverse events (AEs) were also assessed. Homeostatic model assessment of insulin resistance (HOMA-IR) = (FBG × FI)/22.5, where FI is measured in μU/ml and FBG in mmol/l. HbA1c, FI, CRP, hsCRP, TNF-α, and IL-6 were measured in the central laboratory in Shanghai (Shanghai Jince Medical Laboratory Co., Ltd.). TNF-α and IL-6 were measured by enzyme-linked immunosorbent assay (ELISA). The following baseline laboratory variables were analyzed at a local laboratory: clinical chemistry (serum or plasma), S/P-creatine, hematology (whole blood), B-hemoglobin, S/P-LDL-C, S/P-triglycerides, B-platelet count, B-glycated hemoglobin, and urinalysis.
Laboratory safety assessments were not routinely conducted during follow-up visits, but if there were serious AEs or potential endpoint events, the investigator may have decided to conduct different evaluations, including local laboratory evaluations. Follow-up checks for abnormal laboratory results were performed under local practices.
Follow-UpBefore enrollment and at week 16, the participants underwent FBG, complete blood count, urinalysis, and electrocardiography. FCBG levels were measured at week 0, 4, 8, and 12. HbA1c, FCBG, 2 h PPBG, and HOMA-IR were assessed at week 0 and 16. FI levels were measured at week 0, 12, and 16. In addition, laboratory tests were conducted for ALT, AST, TC, TG, FFA, HDL-C, LDL-C, CRP, Hs-CRP, TNF-α, and IL-6 at baseline, week 12 and 16.
Statistical AnalysisThe sample size was calculated using an estimated primary outcome, with an efficacy in the control group of 46.88%, and 66.03% in the test group (HbA1c ≤ 6.5%). In total, 136 participants in each group were required to achieve 90% power and an α level of 0.05 (two-tailed t test). A total of 152 participants were planned to be assigned to both groups to allow for 10% dropouts. The mean and 95% Confidence Interval (CI) were reported for the primary and secondary outcomes, which had an approximately normal distribution. The median and interquartile range (IQR) were reported for outcomes with skewed distribution. Student’s t test and nonparametric tests were used for comparison of means. Chi-square test and Fisher’s exact test were used for contingency table comparison. Both full analysis set (FAS), where data from all participants randomized and at least one time treated were included, and per-protocol set (PPS) analyses, where data exclusively from patients who completed the treatment as initially assigned were incorporated, were performed for sensitivity analysis. The FAS analysis reflects real-world conditions but might introduce bias. In contrast, the PPS analysis offers a more precise evaluation of drug efficacy but forfeits the advantages of randomization. The purpose of employing both analyses is to gain a more comprehensive understanding of the efficacy of drugs in real-world settings. The last observation carried forward method was used for missing data input. PPS analysis was performed on those participants with a > 80–120% compliance rate and an HbA1c value recorded at week 16. Statistical analyses were performed using STATA (Ver. 16.0) and SAS (Ver. 9.4). All statistical tests used in the study were two-sided and considered to be significant if P < 0.05.
留言 (0)