
記住我




Genotype, age at symptom onset and therapies used in our cohort are shown in Table 1. Briefly, 10/12 patients displayed biallelic mutations in C1QA, one in C1QB and one in C1QC. Disease onset occurred during the first years of life (median: 18 months, range: 1–72 months). Patients received multiple immune suppressive therapies (n = 12/12), fresh-frozen plasma transfusion (n = 2/12), plasma-exchange (n = 1/12) or stem-cell transplantation (n = 2/12). After a median follow-up period of 83.5 months (range 48–168), 4/12 patients had died, and another 7/12 had developed mild to severe neurological sequalae.
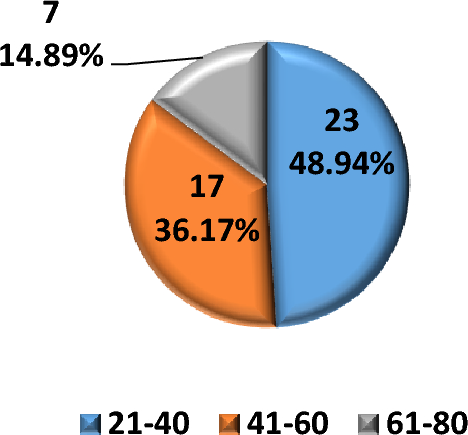
Table 1 Genotype, age at onset, treatment, outcomes and duration of follow-up of n = 12 patients with C1QDef in our cohortClinical manifestations and the auto-antibody profile of patients in our cohort are shown in Fig. 1A. Most patients demonstrated mucocutaneous manifestations (11/12) such as malar rash, oral ulcers, urticarial, vasculitic or pustular (Sweet’s syndrome) rash and alopecia. CNS involvement was recorded in 11/12, encompassing: basal ganglia calcification, CNS vasculitis, moyamoya disease, encephalitis involving the basal ganglia, cerebral atrophy and pachy-meningitis (Fig. 1B-E). By contrast, renal disease and major infections were rare (2/12). Most patients tested positive for ANA (anti-nuclear antibodies) and anti-Ro antibodies (10/12 and 9/12, respectively).
Fig. 1
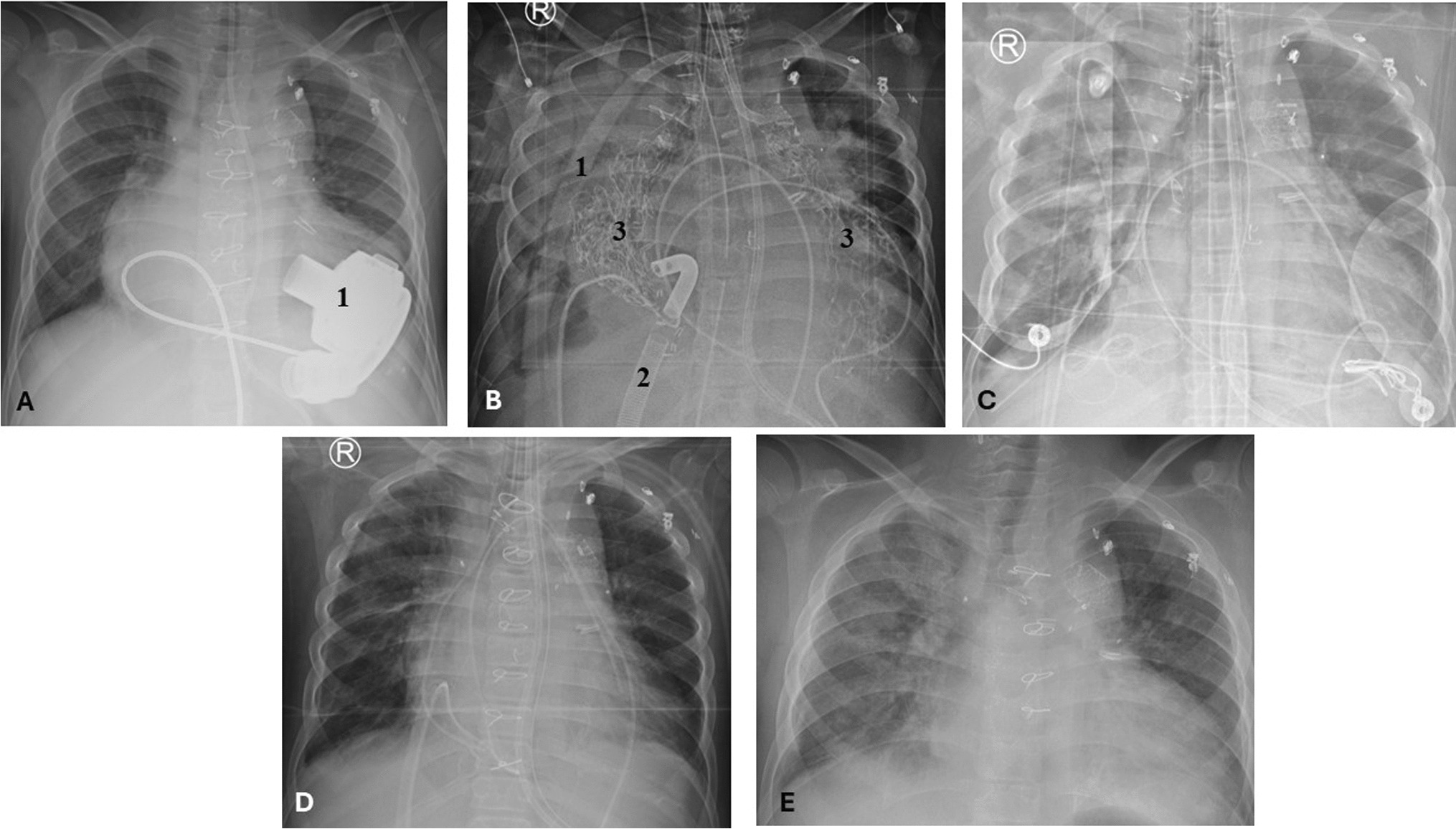
Clinical Features of patients with C1QDef in our cohort. A For each patient, presence (dark) or absence (grey) of the following features is indicated: Anti-nuclear antibody (ANA- specific antibodies are written in the cell if present), mucocutaneous (MC), renal (R), major infection (MI) or central nervous system (CNS) involvement. Black and grey boxes indicate, respectively, the presence or absence of disease. Details of CNS involvement are given in the last column. B CT-scan of patient AGS412 showing basal ganglia calcification. C MRI of patient AGS2522.2 showing encephalitis with signal abnormalities in the basal ganglia and thalami (MRI sequence T2 FLAIR). D MRI (T2W) of patient AGS1000 at relapse of CNS inflammation, showing diffuse enlargement of the left basal ganglia, caudate nucleus and thalamus, with mass effect. E MRI (T2W) of patient AGS1000 two months after (D), showing significant reduction in the size of the lesion and perilesional oedema, and post biopsy changes
Elevated Type 1 Interferon Signature in C1Q DeficiencyWe assessed ISG expression in the whole blood of 10 patients, recording an elevated expression in all patients (Fig. 2A). The interferon signature was found to be in the range of the canonical monogenic interferonopathies Aicardi-Goutières syndrome and STING-associated vasculopathy of infancy (Fig. 2B). In addition, we performed IFNα protein measurement using SIMOA in serum and CSF of two patients with CNS involvement. Both patients (AGS2522.2 and AGS3489, Fig. 1A) displayed elevated serum and CSF IFNα protein levels (223,967.0 and 88,325.8 fg/ml, respectively for patient AGS2522.2; 2468.2 and 159.2 fg/ml, respectively for patient AGS3489) (healthy levels < 10 fg/ml in both serum and CSF).
Fig. 2
ISG expression in peripheral blood of C1QDef. A Expression of 6 ISGs in peripheral blood of n = 9 C1QDef patients compared to controls. ISG expression was determined either by qPCR (n = 8 patients), or NanoString (n = 1 patient). Age at sampling (years) and Interferon score are shown next to each patient ID. B Interferon score of n = 426 samples from canonical monogenic interferonopathies (results are grouped by mutant genotype), n = 79 controls and n = 9 C1QDef patients. Black: controls, red: elevated IS, blue: patient with IS in the range of controls. Whiskers show mean ± error of samples analysed using qPCR. Of note, ISG expression data of patient AGS3489 are not shown here (see Methods)
Clinical and Biological Effects of HSCTTwo patients underwent HSCT. Because of refractory CNS vasculitis, patient AGS1000 was first transplanted using tissue from a mismatched parent (i.e. carrier of one C1Q pathogenic variant). A low stem cell dose led to associated low donor chimerism and undetectable C1Q serum level, but normalized CH50 assay. Two years after HSCT she presented with a relapse of severe CNS inflammation involving the basal ganglia (Fig. 1D). Brain biopsy showed features of vasculitis (presence of mixed B and T cell perivascular and diffuse infiltrates, and fibrinoid necrosis) (Fig. S1). She was treated with high doses steroids, rituximab and mycophenolate mofetil, leading to marked improvement on MRI (Fig. 1E). A second transplantation using a matched unrelated donor was performed. The patient died shortly thereafter from disseminated fungal infection. The second patient (AGS3726) was transplanted using a matched unrelated donor in the context of severe mucocutaneous disease. At last visit (one month after HSCT), the patient was doing well, with normalized CH50 and C1q levels. Transcriptomic data pre/post HSCT were available for the two patients. HSCT significantly reduced ISG expression to the range of controls (Fig. S2).
Clinical Effects of JAK Inhibition in Three Patients with C1Q DeficiencyThree patients in our cohort were treated with JAK-inhibition, with differing outcomes. Patient AGS3489 was started on baricitinib 4 mg/d at age 16 years in the context of active disease despite moderate dose steroids, mycophenolate mofetil and rituximab (see Table S1 for details). Before baricitinib was started, he displayed cutaneous vasculitis, alopecia (Fig. 3A-B), persistent non-infectious pachy-meningitis (Fig. 3E-F), refractory focal epilepsy and elevated expression of ISGs (interferon score = 14.4, normal < 2.7; data not shown in Fig. 2 as performed in another laboratory). After 9 months on baricitinib there was a marked improvement in cutaneous disease (Fig. 3C-D), so that steroids could be tapered from > 12 mg methylprednisolone daily to 6 mg daily. Interestingly, therapy also seemed to improve the associated CNS disease, as signs of pachy-meningitis resolved on MRI (Fig. 3G-H) and focal seizures decreased from weekly to three-monthly crises (with concurrent adaptation of anti-epileptic therapy).
Fig. 3
Clinical and radiological effects of JAKi in a patient with C1QDef. A-D Clinical picture of the face of patient AGS3489 and foot 2 weeks before (A-B) and 9 months after (C-D) initiation of treatment with baricitinib. The complete list of therapies at each time point is given in Supplementary Table 2. E–F Cerebral MRI (T1 sequence with contrasts) of patient AGS3489 3 months before initiation of baricitinib showing cerebral atrophy and pachymeningitis (arrows indicate meningeal thickening with enhancement). G-H Evolution of MRI after 6 months of baricitinib showing improvement in meningeal thickening and enhancement. Of note, a subdural hematoma appeared after lumbar puncture
Patient AGS2522.2 was started on baricitinib 4 mg/d at age 7 years in the context of refractory membranoproliferative glomerulonephritis despite mycophenolate mofetil, steroids and hydroxychloroquine. Under therapy with baricitinib, steroids and hydroxychloroquine, renal disease worsened and severe diffuse encephalitis developed, requiring plasma-exchanges and cyclophosphamide therapy. The patient died of renal and digestive haemorrhage after a renal biopsy in the context of severe, uncontrolled disease (7 months after baricitinib initiation).
Patient AGS1969 has been treated with tofacitinib 2.5 mg twice daily for two years, with only moderate clinical improvement of skin and minimal CNS involvement (hyperreflexia).
Characteristics of 77 Previously Described Patients with C1QDefWe reviewed previously reported cases of C1QDef, focusing only on genetically confirmed cases. We identified 66 such patients in total (n = 32 from previous reviews, n = 34 published cases in 19 publications since 2011). We then combined these data with those of the patients that we report here (n = 12 patients including AGS412 who was described by Troedson et al. [12]) (Table S2).
Regarding genotype, most patients were homozygous (only 5.2% were compound heterozygous), consistent with a high rate of reported consanguinity. Variants were seen in C1QA, CQ1B, and CQ1C in 55.8%, 15.6%, and 28.6% of the 77 patients, respectively. The most frequent variants are shown in Fig. 4A, which were recorded across different ethnic backgrounds (Table 2, Fig. S3), thereby suggestive of mutational hotspots rather than founder effects.
Fig. 4
Review of clinical features and genotypes of n = 77 published cases with genetically confirmed C1QDef. A Most frequent variants reported in C1QDef (73 homozygous and 4 compound heterozygous individuals). B Rate of ANA positivity (ANA), mucocutaneous manifestations (MC), major infections (MI), CNS (CNS) and renal involvement in C1QDef. The absolute numbers of cases with data available for each feature is shown on the right. C Venn diagram showing the co-occurrence of cardinal features of C1QDEF in n = 68 patients with data available for all cardinal features. Only 3 patients were diagnosed without any of these features
Table 2 Three most frequent variants associated with C1QDef reported in the literatureReview of all 77 C1QDef cases confirmed a high frequency of mucocutaneous manifestations (present in 87.5% of patients) (Fig. 4B). CNS involvement was reported in 36.2% of patients; cerebral MRI/CT results were described in 17 of those patients, with 9 (52.9%) demonstrating basal ganglia or deep grey matter involvement. Renal disease and major infections were reported in 23.9% and 37.5%, of patients respectively. ANA titres were positive in 90.8% (59/65) of patients. Although techniques have evolved over time and may vary across laboratories, we analysed the most frequent ANA specificities. At least one ANA specificity was reported in 88.1% (52/59) of patients: Ro in 47.5% (28/59), Sm in 42.4% (25/59), RNP in 32.2% (19/59), and native DNA/dsDNA in 18.6% (11/59). The rate of antibody positivity against native DNA/dsDNA was possibly higher in patients with renal involvement than in those without (7/17 = 41.2% vs 4/54 = 7.4%, p = 0.0027), although this result should be interpreted with caution since more patients in the non-renal groups had missing ANA specificity data (n = 11/54 vs 1/17).
We then analysed the co-occurrence of the major clinical manifestations (mucocutaneous / renal / CNS involvement and major infections) in n = 68 patients with data for these systems (Fig. 4C), finding the most common phenotypes to be: mucocutaneous involvement without other cardinal features (27.9%), and CNS and mucocutaneous involvement (16.2%).
We noted a change in the reported frequency of some features over time (Fig. S4A). Thus, CNS involvement was reported in 20.7% (6/29) up to 2011, and in 47.6% (20/42) since then, with the reporting of severe infections falling from 45.5% (18/33) to 24.4% (10/41) (Fig. S4B-C). By contrast, rates of reported renal and mucocutaneous involvement remained stable.
留言 (0)