
記住我






This is a randomized, double-blind, placebo-controlled, single ascending dose (SAD) and multiple ascending dose (MAD) phase I study conducted in the Third Hospital of Changsha in China from January 7, 2023 to April 21, 2023. The Human Interferon Alfa-1b Inhalation Solution (GB05) was manufactured according to FDA guidelines and supplied by Kexing Biopharm Co., Ltd [16].
The study protocol and informed consent forms were approved by the Institutional Review Board (IRB)/Independent Ethics Committee (IEC) of the Third Hospital of Changsha (Approval number CS3-2022EC-065). This study was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments. All participants provided written informed consent for this study.
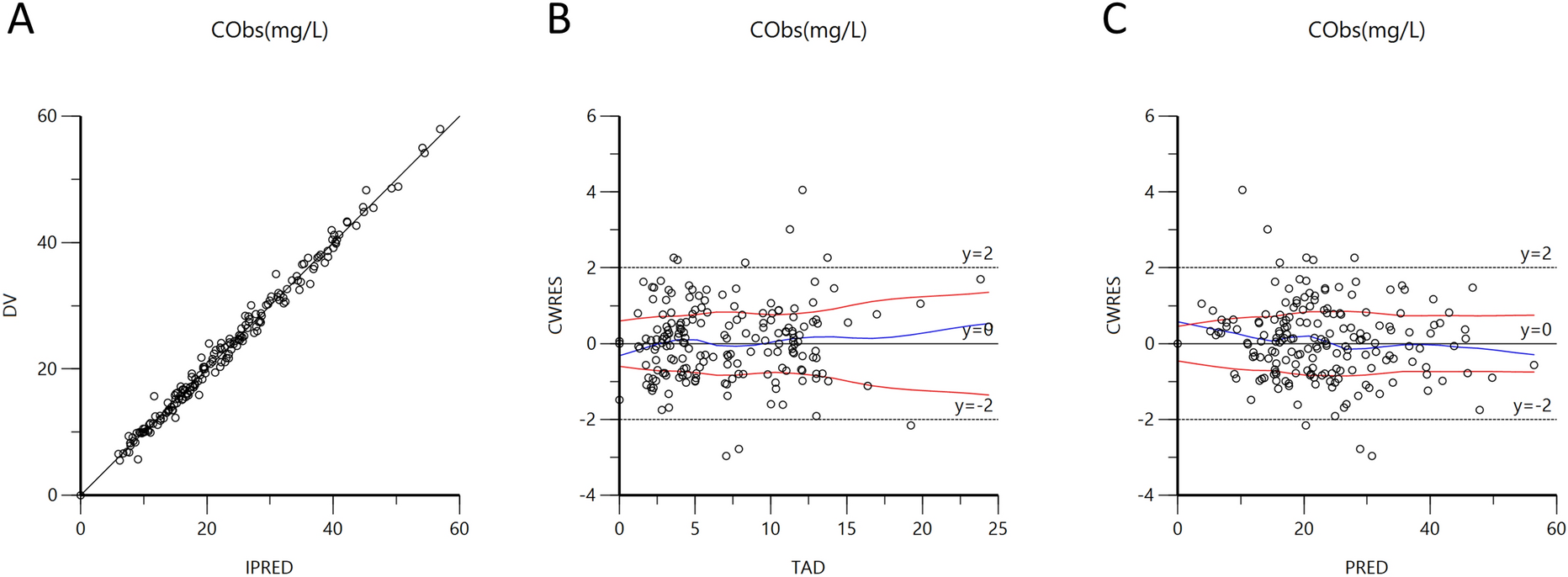
As a first-in-human clinical trial, the dose range settings of GB05 were based on the no observed adverse effect levels (NOAELs) originating from the long-term GB05 toxicological studies conducted in cynomolgus macaques, in which the NOAEL was 14.602 μg kg−1 day−1 (126,100 IU kg−1 day−1). On the basis of preclinical dose studies, four groups were set up for the SAD study: the proposed initial single dose was 0.2 million IU, followed by 0.6, 1.2, and 1.8 million IU. For the MAD study, 1.2 million IU and 1.8 million IU groups were then given multiple consecutive doses after the single dose, i.e., 1.2 million IU bid (daily dose of 2.4 million IU), 1.8 million IU bid (daily dose of 3.6 million IU) (Fig. 1a).
Fig. 1
Study designs. a The scheme of dose escalation design of SAD (blue box) and MAD (purple box). b Intervention schemes. SAD single ascending dose, MAD multiple ascending dose, D day
Eligible volunteers were healthy Chinese adults aged 18–45 years with a normal body mass index (19.0–26.0 kg/m2). Key exclusions from the study included volunteers who have a history within 3 months of any surgery or planned surgery during the trial, respiratory system diseases, ocular diseases, thyroid-related diseases, drug allergies, cannot tolerate nebulized inhalation, and other criteria that would affect the absorption, distribution, metabolism, and excretion of drugs.
Randomization and MaskingThe randomization of GB05 and placebo groups was performed by an independent statistician who was not involved in the trial. Random numbers were generated for volunteers with a randomization sequence generator called PROC PLAN of SAS 9.4 (SAS Institute Inc, Cary, NC). In this study, eligible volunteers were randomized respectively among the four dose groups, with a ratio of 4:1 between GB05 and placebo in all groups.
The investigational drug for each volunteer was packaged in sealed, identical boxes to conceal the intervention allocation. The independent statistician affixed the appropriate number to the package according to the drug coding table. The independent statistician will keep random number tables concealed until the end of the study.
InterventionIn this study, both GB05 and placebo solutions were provided by Kexing Biopharm Co., Ltd., produced following GMP standards. For the first and second groups, each volunteer was given the corresponding dose of nebulized GB05 or placebo solution in the morning of day 1, and only a single dose was administered during the trial. For the third and fourth groups, each volunteer was administered two doses with the corresponding dosage of GB05 or placebo solution at an 8-h interval each day from day 4 to day 7, and a final dose in the morning of day 8. In general, the MAD groups were administered for five consecutive days, with a total of nine doses during the trial (Fig. 1b).
Safety and Tolerability AssessmentsSafety and tolerability of GB05 were assessed on the basis of adverse events (AEs)—significant clinical changes in the vital signs, physical examination, 12-lead electrocardiogram (ECG), clinical laboratory tests (routine blood test, blood chemistry, coagulation function, routine urine test, thyroid function, complement C3), and pulmonary function tests after administration. All AEs were assessed and classified during the trial according to Common Terminology Criteria for Adverse Events 5.0 (CTCAE 5.0). AEs were monitored throughout the study and were evaluated by the investigator for the association with GB05.
Dose escalation stopping criteria were set according to CTCAE 5.0. Dose escalation for the group would be stopped (a) if the same treatment-emergent adverse event (TEAE; AEs that happen within the administration period), greater or equal to level 2, happens among more than half of the volunteers, or (b) if any TEAE, greater or equal to level 3, happens among more than one-third of the volunteers in the same dosage group. If any serious adverse event (SAE; important medical events that occurred after treatment, such as death, life-threatening, permanent or serious disability or loss of function) related to treatment occurs in any volunteer, administration for the volunteer would be terminated.
The drug will be considered tolerated if none of the criteria were met during the study.
Pharmacokinetic AssessmentsSerial blood samples for PK evaluation were collected from all volunteers at specified time points. For SAD groups, samples were taken 0.5 h pre-dose on day 1 and at 0, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24, and 48 h after administration. For MAD groups, we specified day 3 as time point 0, samples were taken 1 h pre-dose on day 6 (1 dose), day 7 (2 doses), and day 8 (1 dose), 1 h pre-first and second dose on, and then at 0, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 24, 48, 120, 144, and 168 h after day 8 administration. Blood samples for PK analysis were collected into labelled 4-mL serum separator tubes and allowed to clot. Samples were centrifuged and supernatants (serum) were stored at − 80 °C.
The bioanalytical analysis for PK samples was performed by United-Power Pharma Tech (Shanghai) Co., Ltd. Serum concentration of IFNα1b was determined using a validated enzyme activity method with a concentration range from 10 to 640 pg/mL, and PK parameters were calculated. Briefly, blank microplates were pre-coated with anti-interferon alpha 1 antibody (Abcam, ab20200). After blocking and washing were performed, diluted serum (1:4) and the positive (Kexing Biopharm), negative, and blank controls were added. After incubation and washing were performed, recombinant anti-interferon alpha 1 antibody (Abcam, ab242952) was added, followed with Peroxidase AffiniPure Donkey Anti-Rabbit IgG (H + L) (Jackson ImmmoResearch, EPR18694-27). TMB (3,3′,5,5′-tetramethylbenzidine) solution was added to each well and incubated for 15 min. The reaction was stopped by adding stop solution to each well and the optical density (OD) at 450 nm was determined with an ELISA plate reader. OD values of each concentration point were collected and processed with the SoftMax Pro 6.5.1 GxP software. The standard curve is generated by Watson LIMS software (Version 7.5) for each plate, using a 5-PL (Marquardt) model with 1/Y weighting factor.
Immunogenicity AssessmentImmunogenicity assessment of GB05 was conducted via anti-drug antibody (ADA) assay. ADA positive samples would then be analyzed for titer and neutralizing antibodies (NAb). The number and percentage of volunteers with positive immunogenicity (ADA and NAb) should be described if the data allowed. For volunteers with positive immunogenicity, the time of onset and duration should be further described.
Study EndpointsThe primary endpoint is safety and tolerability, assessed by AEs and incidence, TEAEs, adverse drug reactions (ADRs, any harmful or unexpected reaction that may be related to GB05 administration), SAEs, and TEAEs that lead to study discontinuation from the study.
The secondary endpoint is PK and immunogenicity. For plasma PK parameters of SAD administration: maximum concentration (Cmax), the time taken to reach the maximum concentration (Tmax), area under plasma concentration–time curve from time 0 to the minimum detectable concentration (AUC0–t), and, if data were available, area under the curve from time 0 to infinity ∞ (AUC0–∞), half-life (t1/2), elimination rate constant (Ke), apparent distribution volume (Vd), mean residence time (MRT), and clearance rate (CL), were calculated.
For plasma PK parameters of MAD administration: steady-state trough concentration (Css_min), steady-state maximum concentration (Css_max), average steady-state plasma concentration (Css_av), and, if data were available, fluctuation coefficient (DF) and fluctuation amplitude [(Cmax,ss − Cmin,ss)/Cmin,ss], were calculated.
For the first and second dose groups, immunogenicity was evaluated within 1 h before day 1 administration, then at day 9 ± 1 day, day 16 ± 1 day, and day 23 ± 2 days. For the third and fourth groups, immunogenicity was evaluated within 1 h before day 1 administration, then at day 18 ± 1 day, day 25 ± 2 days, and day 32 ± 2 days.
Statistical AnalysesThe statistical methods of this study are descriptive statistics rather than hypothesis tests, and the sample size was chosen on the basis of common tolerability or PK study cohort. Safety set (SS) refers to all volunteers randomized and who received investigational drugs with records of safety assessments. Full analysis set (FAS) refers to all randomized volunteers who received the investigational drug. PK concentration set (PKCS) refers to all volunteers randomized and who received investigational drugs with at least one post-dose plasma drug concentration data collection during the trial. PK parameter set (PKPS) refers to all volunteers randomized and who received investigational drugs with at least one effective PK parameter during the trial.
PK analysis was performed on the basis of the non-chamber ventricular model with WinNonlin software (Certara USA Inc, version 8.2 or above), and other analyses were performed using SAS (version 9.4 or above) software. AEs during the study were coded according to the ICH Medical Dictionary for Regulatory Activities (MedDRA24.1 or above version). The data were expressed as mean ± standard deviation or as numbers with proportions. P values < 0.05 were regarded as statistically significant.
留言 (0)