
記住我


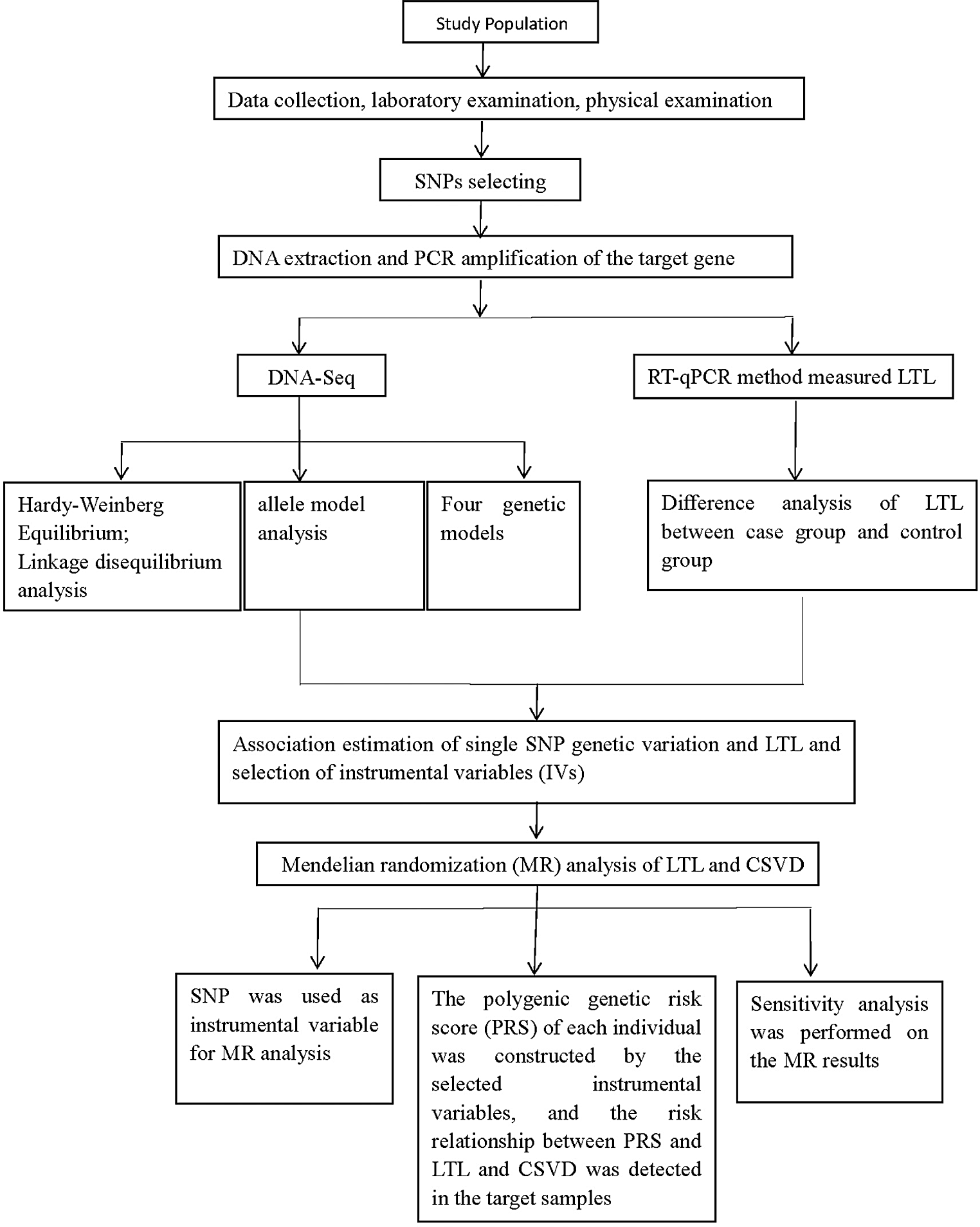
Prospective cohort study of patients residing in Denmark with a suspected or already diagnosed hereditary anemia (Fig. 1). The study was approval by the Ethical Committee of the Capital Region of Denmark (H-21064560) and the Danish Data Protection Agency (P-2021-736). Patients provided informed consent to participate.
Fig. 1
Flowchart of the Danish Hereditary Anemia (DAHEAN) study. Any patient suspected of or diagnosed with hereditary anemia may be included. Consent is necessary for participation. Whole genome sequencing (WGS) may be performed if clinically indicated. When necessary clinical and paraclinical data has been gathered, patients are discussed at the monthly national anemia boards, which provides diagnostic and therapeutic recommendations to the treating physician
Patients were included from participating clinical departments (Table 1). Patients with existing hereditary anemia diagnoses were included at the physician’s discretion to validate, refine, or change previous diagnoses using WGS, identify additional genetic variants affecting disease management, and enhance understanding of hereditary anemias. This approach aims to open new avenues for treatment and care by integrating advanced genetic insights. Diagnostic procedures were only performed when relevant and consequently, not all analyses are available on all patients.
Table 1 Participating departmentsData capturePatient data were collected using REDCap (Research Electronic Data Capture) electronic data capture tools hosted by the Capital Region of Denmark [18, 19]. REDCap is a secure, web-based software platform designed to support data capture for research. Each department were allocated to a data access group in which they can only access their own patients. Only administrators have access to patient information across sites.
WGS raw data were stored by Danish National Genome Center (NGC; https://eng.ngc.dk) according to standard operating procedures.
Incidental genetic findingsPseudopanels were used for extensive genetic testing, which minimized the risk of detecting pathogenic germline variants unrelated to anemia in patients. If such variants were detected, patients were offered a referral to the genetic counseling services at their local Department of Clinical Genetics.
Study subjectsPatients were recruited from hematological, pediatric, and clinical genetic departments and offered inclusion through their local physician. Inclusion criteria were broad: (1) All ages, (2) Signed informed consent from patient or their legal guardian (consents currently available in Danish and English), (3) Suspicion or verified diagnosis of a hereditary anemia. There were no exclusion criteria. Criteria were intentionally left very broad to facilitate that any patients that might benefit from comprehensive evaluation for hereditary anemia could be included. Consequently, cohort composition very much depended on the referring physician’s preference, which will likely also develop over time as physicians – hopefully – experience benefit of including patients.
As pathogenetic PIEZO1 mutations can cause a membranopathy - patients evaluated genetically for JAK2-wildtype erythrocytosis were included in the cohort. Additionally, genetic disorders of iron metabolism may also impair erythropoiesis and patients suspected of these were also eligible for this cohort.
Clinical informationRelevant clinical information was gathered by the patient’s physician and typed into a secured electronic database (see above). Information included name, social security number, relevant medical history, blood works, blood transfusion data, radiology, family medical history, and ethnic origin. Ethnicity was essential to genetic analysis as frequency of gene polymorphisms vary in different ethnic populations and thus influence the evaluation of genetic variants. Data were, whenever possible, captured in a structured manner using standardized tools such as human phenotype ontology (https://hpo.jax.org) and SNOMED (www.snomed.org).
Genetic analysesWhole genome sequencing (WGS) was performed by the NGC according to NGC standard operating procedures. Originally, WGS was envisioned as a last resort for cases of hemolytic anemias that remained unresolved after exhausting the standard diagnostic pathway (Fig. 2). Nonetheless, the adherence to this protocol was not enforced, granting the referring physician the autonomy to decide on the use of WGS based on their clinical judgment.
Fig. 2
Diagnostic Flowchart for Hereditary Anemia. Structured approach for adding whole-genome sequencing (WGS) to the diagnostic algorithm. diagnosing hereditary anemia. Abbreviations: AIHA: Autoimmune Hemolytic Anemia, DAT: Direct Anti-globulin Test, EMA: Eosin-5-maleimide, G6PD: Glucose-6-Phosphate Dehydrogenase, MCV: Mean Corpuscular Volume, PNH: Paroxysmal Nocturnal Hemoglobinuria, WGS: Whole Genome Sequencing
WGS was done using Illumina PCR-Free library preparation and Illumina platform sequencing. Bioinformatic processing was based on a standardized and continuously updated pipeline. Alignment of sequencing reads to the hg38/GRCh38 reference genome was done using BWA [20]. Single nucleotide variant (SNV) calling was performed with GATK (following best practices) [21]. Structural variant (SV) calling was performed using a combination of several tools: Manta [22], Delly2 [23], Lumpy [24] and CNVnator [25]. Variants were annotated and filtered using VarSeq (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com). These analyses required consent to comprehensive genetic analyses (available from www.anemia.dk). Although used in most patients, WGS was not a prerequisite for participation in the study.
More than 160 genes implicated in hereditary anemias were included in a continuously updated gene panel (Supplementary Table 1), that was used for in silico filtering of variants. However, novel anemia associated genes could also be searched for and identified. This especially pertained to patients with a family history of anemia of unknown origin or patients with unusual phenotype. Gene prioritization of anemia associated genes is done using the inbuilt VarSeq algorithm PhoRank.
Variant interpretation and classification was done according to recommendations from the American College of Medical Genetics (ACMG) [26] with inclusion of refinements to the guidelines recommended by Clinical Genome (ClinGen) Resources Sequence Variant interpretation subgroup (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/) and relevant ClinGen expert panels. The molecular genetic effect of variants was evaluated using VarSeq and the integrated software Alamut Visual Plus (SOPHiA GENETICS, USA). Classification was based on information from relevant clinical (e.g. NCBI ClinVar, HGMD [27]) and population databases (e.g. gnomAD [28]), extensive searches for prior reports in the medical literature, and in silico predictions using REVEL [29] for missense variants and a combination of MaxEntScan [30] and SpliceAI [31] for splice altering variants. Online Mendelian Inheritance in Man (OMIM) nomenclature was used to subtype genetic diseases.
In selected cases, other genetic techniques such as array CGH, Sanger sequencing, and targeted sequencing of specific genes [32] was employed.
Functional assaysFunctional assays were used to diagnose hereditary anemias and to verify the pathogenicity of genetic variants found.
Protein quantificationMass-spectrometry based proteomics and metabolomicsAs a pilot project, blood from selected patients investigated for hereditary anemias were subjected to mass-spectrometry based proteomics at the Department of Clinical Biochemistry at Bispebjerg Hospital [33] and metabolomics at the University Medical Center Utrecht, The Netherlands [34].
Protein and peptide measurementsHemoglobin fractions were routinely quantified by high-pressure liquid chromatography (HPLC) [35, 36] at Aarhus University Hospital and Rigshospitalet. Specific protein quantification could be performed by Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE), Western blotting, and Enzyme-Linked Immunosorbent Assay (ELISA). Assays of erythropoiesis and hemolysis including erythroferrone, GDF15, soluble transferrin receptor, and hepcidin were available [37] if relevant. Hemoglobin stability was assessed by isopropanol precipitation test [38].
CytologyBlood marrow and peripheral blood were assessed according to standard procedures at the local department of pathology [39]. Central review at the Department of Pathology at Rigshospitalet was often utilized.
MembranopathiesRBC membrane were studied by standard diagnostic procedures including osmotic gradient ektacytometry (RR Mechatronics) [11, 32, 40] and flow cytometry including Eosin-5-maleimide (EMA)-binding test [32, 36, 40].
Functional tests of RBC ion channels were experimental and lacked standardization. With this cohort, we aim to validate novel assays, including one automated patch-clamp [41].
EnzymopathiesEnzymatic assays were performed to verify enzyme deficiencies. These included measurement of pyruvate kinase [42] and glucose-6-phosphate dehydrogenase (G6PD) activity. Enzymatic assays unavailable in Denmark were performed at the EuroBloodNet laboratory at the University Medical Center Utrecht, The Netherlands.
Hemoglobin oxygen affinityOxygen affinity of hemoglobin was assessed by local arterial blood lactate (ABL) analyzer or more detailed methods such as HEMOX [43]. For when patients with sickle cell disease are enrolled, Oxygenscan [44] is available for phenotype assessment.
Autoimmune assaysAutoimmune assays include direct antiglobulin test, cold agglutinin titers, Donath-Landsteiner test. Screening tests were typically performed locally, but more sensitive and specific flowcytometric tests were available at the Department of Clinical Immunology at Rigshospitalet [45].
Statistical considerationsDue to the exploratory nature of the study, the study did not aim to reach a specific number of patients and a power calculation was therefore not needed. Inclusion of 50–100 patients per year was expected.
European collaborationPatients identified with a rare hereditary anemia were offered inclusion in the Rare Anemia Disorders European Epidemiological Platform (RADeep; https://www.radeep.eu) to help map prevalence, increase understanding of disease phenotype, and promote translational research in rare anemias.
Patients who remained diagnostically or therapeutically unresolved after a DAHEAN conference could be assessed via the Clinical Patient Management System of ERN-EuroBloodNet (https://cpms.ern-net.eu) to provide specialized, expert medical care on a European level.
留言 (0)