
記住我








A 60-year-old male presented to the local accident and emergency department feeling generally unwell with shortness of breath, icteric sclera, and jaundice. Initial full blood count (FBC) showed pancytopenia, haemoglobin 100 g/L, white cell count 1.3 × 109/L, platelet count 30 × 109/L, and an absolute neutrophil count (ANC) of 0.4 × 109/L. A blood film performed on peripheral blood (PB) revealed a leucoerythroblastic picture and pancytopenia. The coagulation screen and kidney function tests were normal. Serum lactate dehydrogenase (LDH) and C-reactive protein (CRP) were 492 U/L and 28 mg/L respectively. The patient had an extensive past medical history including non-ST segment elevation myocardial infarction (NSTEMI), obesity, type 2 diabetes mellitus (T2DM), hypertension, atrial fibrillation, and recurrent cellulitis. A bone marrow aspirate and trephine was performed which was grossly hypercellular, almost entirely replaced with medium/large blasts/promyelocytes with variable amounts of cytoplasm containing fine azurophilic granules, some with budded cytoplasm and many with a folded nucleus (Fig. 1A and B). These morphologic features raised suspicion of APL. Immunophenotyping analysis was performed and indicated that the blasts/promyelocytes were CD34-, HLA-DR(weak) + , MPO + , CD64 + , CD13 + ,CD33 + , and CD117 + myeloid lineage cells. As a consequence of what appeared to be classic APL morphology and immunophenotyping results, the patient was commenced on ATRA 45 mg/m2, awaiting urgent FISH and real-time quantitative PCR (RQ-PCR).
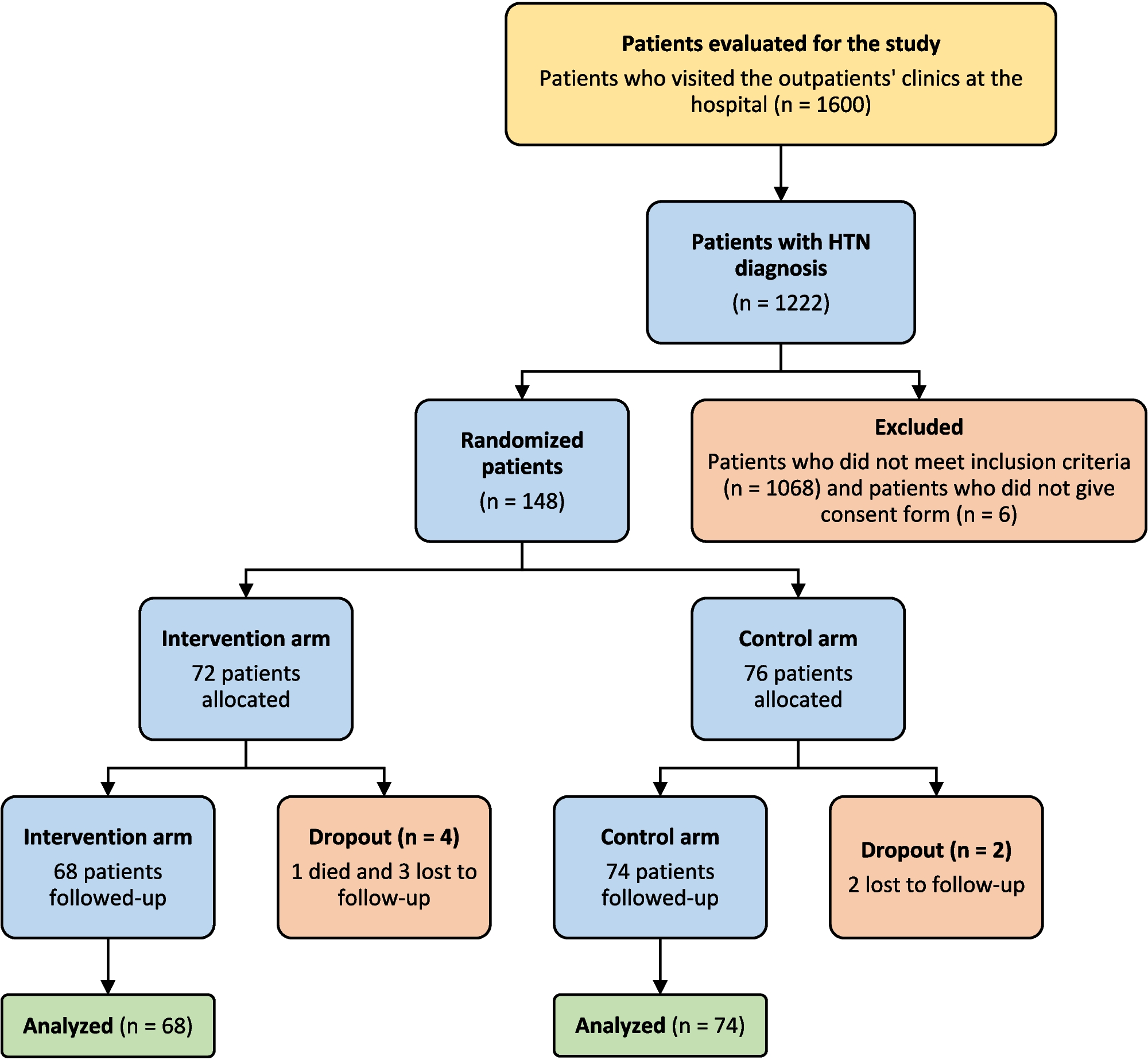
Fig. 1
Initial bone marrow aspirate morphology and cytogenetic analysis of the patient. A and B Bone marrow aspirate showing hypercellularity and a population of medium-sized blasts with budded cytoplasm (black arrow), nuclear folding (yellow arrow), and some with Auer rods (red arrow). Pictures taken with oil at × 50 magnification. C Interphase nuclei showing one intact copy of RARα and two copies of the 5’ portion of the RARα probe. D Metaphases showing two 5’RARα signals hybridised to the der(17). E Isochromosome for the short arm of chromosome 17 and a derivative chromosome 17 which includes two copies of the long arms of chromosome 17. F Sanger sequencing of the STAT5b-RARα fusion. The Sanger sequencing at the breakpoint site is depicted by the black line
The patient subsequently tested negative for a PML::RARα rearrangement by both FISH (Fig. 1C) & RQ-PCR, following the protocol described by Europe Against Cancer (EAC). There was also no evidence of MECOM or KMT2A rearrangements. Chromosome analysis (Fig. 1E) of bone marrow cell cultures showed a karyotype consisting of 47 chromosomes including an isochromosome for the short arm of chromosome 17. There was also a derivative chromosome 17 which includes two copies of the long arms of this chromosome. ATRA treatment was discontinued at this point however subsequent FISH analysis, this time using an Vysis LSI RARα dual colour break-apart probe, displayed one intact copy of RARα along with two copies of the centromeric portion of the RARα probe. These copies were confirmed by metaphase FISH to be located on the der(17)—ATRA was therefore resumed pending further investigation. Three copies of TP53 were also identified. Based on karyotyping and FISH analysis, the results therefore represented a variant RARα rearrangement with an unknown cryptic partner gene.
In order to identify this cryptic partner, the sample was sent for myeloid next generation sequencing (NGS). DNA libraries were prepared using the KAPA Hyper Plus Kit (Kapa Biosystems) and sequenced using a Myeloid NGS assay covering common sequence, structural and copy number variations in genes involved in myeloid neoplasms. Alignment, de-duplication, and variant calling was performed using the MyeloidTS_WF bioinformatics pipeline v1.1. This panel uncovered a cryptic inv(17) or del(17)(q21.2q21.2) STAT5b e15 (NM_012448.4)::RARα e3 (NM_000964.4) variant. The sample was also sent to Newcastle Genetics laboratory, Newcastle upon Tyne, for an RNA fusion assay using the Illumina TruSight panel and confirmed by Sanger sequencing (Fig. 1F). Therefore, based on these genomic findings, the favoured classification according to the 2022 WHO classification is “APL with a variant RARα translocation” [1]. This is also in keeping with the International Consensus Classification [24].
The patients’ disease was judged to be resistant to ATO or ATRA, and due to extensive co-morbidities, he was not a candidate for intensive chemotherapy (ICT). Therefore, he was commenced on five cycles of low dose cytarabine (LDAC), 20 mg/m2 given subcutaneously once daily on days 1–10 of each 28 day cycle, beginning on cycle 1 day 1, and venetoclax (Ven) 100 mg from day 4 to day 28 orally with an azole after dose escalation from days 1–4. The bone marrow biopsy following cycle 1 was hypocellular with no excess of blasts/promyelocytes, and the patient was commenced on daily granulocyte colony-stimulating factor (G-CSF). Morphological remission (MR) was achieved at this point, demonstrating a clear sensitivity to a LDAC/Ven regimen. However, STAT5b::RARα mutant transcripts were still detectable by molecular minimal residual disease (MRD) and flow MRD was also positive. Cycle 2 was well tolerated. However, venetoclax dosage was reduced to 14 days as a consequence of prolonged cytopenias. Cytogenetic remission was achieved following this cycle. The patient proceeded with cycles three and four respectively with a delay in cycle five due to cytopenias and probable cellulitis. A subsequent bone marrow aspirate unfortunately revealed progressive disease morphologically (approximately 33% blasts) and continued MRD positivity. The patient was transferred to ICU with neutropenic sepsis with gram-positive cocci in culture and was treated with multiple broad spectrum antibiotics. The patient continued to deteriorate with persistent pyrexia and pancytopenia requiring transfusion support and died in ICU.
留言 (0)