
記住我



Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus (Tay et al., 2020; van Eijk et al., 2021) that has caused a pandemic of acute respiratory syndrome in humans following the outbreaks of Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome Coronavirus (MERS-CoV; Zhou et al., 2020). Since the outbreak of COVID-19, over 600 million people have been confirmed to have tested positive for the virus, and more than 6 million patients have died. SARS-CoV-2 shares all typical structural features of other coronaviruses (Liang et al., 2020). However, compared with SARS-CoV, SARS-CoV-2 has certain structural changes that render it more infectious.
Similar to SARS-CoV, SARS-CoV-2 enters the cells by binding to its primary functional receptor, angiotensin-converting enzyme 2 (ACE2; Hashimoto et al., 2012; Varga et al., 2020), which is widely distributed in epithelial cells and in endothelial cells of tissues and organs throughout the body (Bourgonje et al., 2020; Yan et al., 2020). Both the tissue distribution of ACE2 and the effect of SARS-CoV-2 infection on the physiological functions of ACE2 determine, to a certain extent, the clinical features of the disease (Bourgonje et al., 2020).
COVID-19 cases present a wide range of clinical manifestations, ranging from mild symptoms such as dry cough, fever, and sore throat to severe acute respiratory distress syndrome (ARDS), multi-organ failure (MOF), and even death. The severity of COVID-19 is associated with the host immune response to SARS-CoV-2 (Huang et al., 2020; Tay et al., 2020). An effective immune response is crucial for viral clearance, while a dysfunctional response leads to viral persistence and excessive production of inflammatory factors, resulting in cytokine storm and disease progression. Therefore, understanding the interaction between SARS-CoV-2 and the host immune system, as well as identifying and characterizing the abnormal immune response induced by the virus, are essential for developing effective therapies.
Different disease stages in COVID-19 should be managed with tailored treatment options based on the pathogenic characteristics of SARS-CoV-2. During the early phase of the disease course, inhibiting viral entry and replication is critical, whereas in the late phase of the disease with high inflammatory status, measures to alleviate or control the inflammatory response should be applied (Schurink et al., 2020; Kim et al., 2021). Here we briefly overview the structural features of SARS-CoV-2 and its interaction with ACE2, elucidate some of the progress in understanding the associations of innate immunity, adaptive immunity and the cytokine storm with the immunopathogenesis of COVID-19, discuss the emerging variants of concern/interest (VOC/I) and several potential therapeutic options, outline the efficacy of different vaccines against SARS-CoV-2 and its variants. We provide insights into the treatment and management of the disease, exploring both individualized treatment strategies and the development of effective vaccines for future public health crises caused by coronaviruses.
2 Structure of SARS-CoV-2The SARS-CoV-2 virus particles exhibit a spherical structure with a diameter of ~65–125 nm, and the spike (S) protein located on the viral surface gives the viruses a crown-like (“corona”) appearance, from which they acquired their name (Liang et al., 2020; Shereen et al., 2020). Enveloped coronaviruses possess the largest single-stranded, positive-sense RNA genome, with a length of approximately 26–32 kilobases (de Wit et al., 2016; Shereen et al., 2020; Walls et al., 2020). Two-thirds of the coronaviral genome encodes non-structural proteins, including RNA-dependent RNA polymerase (RdRp), proteases and helicase, which are responsible for viral replication; the 3′-end of the genome encodes the four main structural proteins of the coronavirus particles, namely the S, membrane (M), envelope (E) and nucleocapsid (N) proteins, as well as several accessory proteins that interfere with the host innate immune response (Pyrc et al., 2006; de Wit et al., 2016).
2.1 Spike proteinThe S protein of SARS-CoV-2 has a trimeric structure and is located on the viral surface, giving the virus a “crown-like” appearance and serving as the major antigen that the host immune response targets (Liang et al., 2020; Shereen et al., 2020; Walls et al., 2020). SARS-CoV-2 infects host cells by binding to ACE2 through the S protein, which consists of two subunits, S1 and S2. The receptor-binding domain (RBD) of S1 binds receptors on the surface of host cells, allowing virus particles to attach to cells, while S2 promotes membrane fusion and the entry of RNA genes (Walls et al., 2020; Wrapp et al., 2020). Although the RBDs of SARS-CoV and SARS-CoV-2 share 72% similarity in their amino acid sequences (Tay et al., 2020), computational modeling and biophysical measurements have shown that SARS-CoV-2 RBD binds to ACE2 with higher affinity than the RBD of SARS-CoV (Wrapp et al., 2020). This may be due to the replacement of Val 404 in the SARS-CoV RBD with Lys 417 (a polar amino acid) in the SARS-CoV-2 RBD, resulting in tighter binding to ACE2 (Yan et al., 2020). However, the SARS-CoV-2 RBD is less exposed than the SARS-CoV RBD and is mostly found in a “lying-down” state that cannot bind to ACE2, which may be one of the immune evasion mechanisms of SARS-CoV-2 (Yuan et al., 2017; Shang et al., 2020). In addition, the S protein of SARS-CoV-2 contains a furin-like cleavage site that is absent in SARS-CoV, which enhances viral fusion with host cell membranes and probably contributes to the increased infectivity of SARS-CoV-2 compared to SARS-CoV (Walls et al., 2020; Wrapp et al., 2020).
2.2 M, E, and N proteinsThe M protein of SARS-CoV-2 exists as a dimer and is the most abundant protein in the virus. It maintains the viral skeleton, transports nutrients across the membrane, promotes the release of nascent viral outgrowth, and plays a central role in viral assembly (Neuman et al., 2011). The E protein of SARS-CoV-2 is an ion channel that is essential for virus-host interactions and is associated with virulence. Moreover, the E protein participates in viral assembly and release (Nieto-Torres et al., 2014; Liang et al., 2020). Finally, the LKR region (also called SR) of the N protein of SARS-CoV-2 is a serine- and arginine-rich sequence that assists in cell signaling processes (You et al., 2005; Hurst et al., 2009; McBride et al., 2014).
2.3 Accessory proteinsThe SARS-CoV-2 genome encodes different accessory proteins, including ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9b, ORF9c, and ORF10, that can play key roles in the viral pathogenesis and immune evasion processes (Zandi et al., 2022). Therefore, these accessory proteins could be considered potential drug targets.
3 Cell entry of SARS-Cov-2 3.1 ACE2 receptorACE2 is a homolog of angiotensin converting enzyme (ACE; Bourgonje et al., 2020), and plays a critical role in the renin-angiotensin system (RAS). ACE2 cleaves angiotensin II (Ang II) into angiotensin 1-7 (Ang1-7), which binds to the G protein-coupled receptor Mas, inhibiting the p38 MAPK and nuclear factor kappa B (NF-κB) signaling pathways, exerting expanded tubulars, anti-inflammatory, and anti-fibrotic effects (de Carvalho Santuchi et al., 2019; Magalhaes et al., 2020; South et al., 2020). The ACE2/Ang (1-7)/Mas receptor axis therefore functions in opposition to the ACE/Ang II/AT1 receptor axis (Bourgonje et al., 2020; Figure 1). ACE2 is the functional receptor for SARS-CoV-2 and SARS-CoV. One study demonstrated that SARS-CoV infection resulted in the downregulation of ACE2, which contributed to disease severity by disrupting the renin angiotensin aldosterone system in a rodent model (Kuba et al., 2005). The same is presumed for SARS-CoV-2, but the downregulation or exhaustion of ACE2 has not been demonstrated in the human SARS-CoV-2 model either in vitro or in vivo.
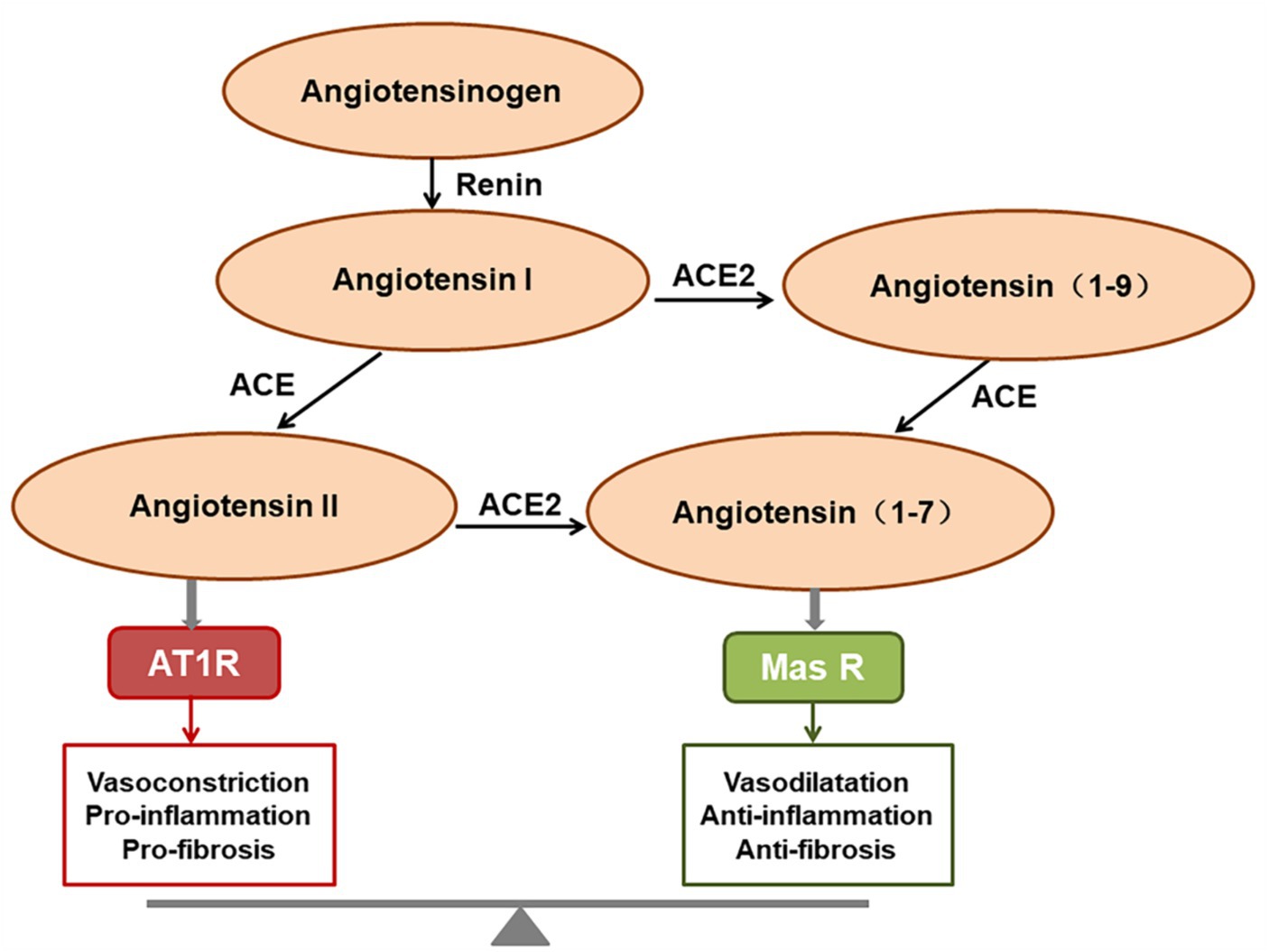
Figure 1. The function of the renin-angiotensin system (RAS). Angiotensin II predominantly exerts its role via activating the AT1 receptor, whereas angiotensin (1-7) [Ang (1-7)] binds to the Mas receptor to exert the opposing effects. The final homeostatic effect of the RAS depends on the balance of the angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/AT1R axis with the ACE2/Ang (1-7)/Mas receptor axis.
3.2 Cell entry mechanisms of SARS-CoV-2SARS-CoV-2 enters host cells through two pathways, namely endocytic pathway and transmembrane serine protease 2 (TMPRSS2)-mediated surface pathway (Nazerian et al., 2021; van Eijk et al., 2021; Zhao et al., 2021). When SARS-CoV-2 binds to ACE2 through the RBD of the S protein, the structure of the S protein changes, allowing TMPRSS2 and cathepsins on the cell surface to cleave the S protein. Subsequently, proteolytic processing at the S1/S2 boundary and the S2 cleavage site removes the covalent connection of the two functional subunits, leading to the dissociation of S1 and facilitating the exposure of the S2 subunits’ domain, which initiates the fusion reaction by inserting of the hydrophobic fusion peptide into the cell membrane (Hoffmann et al., 2020; Walls et al., 2020; Wrapp et al., 2020). After binding, the virus becomes enveloped within endosome formed by the cell membrane and enters the cell through endocytosis. The early endosomes subsequently mature and acidify to form late endosomes, which ultimately develop into lysosomes. In lysosomes, lysosomal cathepsins facilitate the fusion of viral particles with the lysosomal membrane. Fusion between viral and cellular membranes forms a fusion pore through which viral RNA is released into the cytoplasm of the host cell for replication (Nazerian et al., 2021; Jackson et al., 2022). SARS-CoV-2 can also enter the cell via the TMPRSS2-dependent direct fusion of the viral envelope with the cell membrane, but the endocytic pathway is considered the main route (Heurich et al., 2014; van Eijk et al., 2021). Additionally, pre-activation of the protein convertase furin enhances the entry of SARS-CoV-2 into certain target cells, especially those with relatively low expression levels of TMPRSS2 and/or lysosomal cathepsins, contributing to increased infectivity (Shang et al., 2020).
4 Immune response against SARS-CoV-2 and immune defects 4.1 Innate immune response 4.1.1 Innate immune dysregulationThe innate immune response provides the first defense against pathogenic microorganisms, and plays an important role in resisting viral invasion and initiating an adaptive immune response. Upon entry into cells, the pathogen-associated molecular patterns (PAMPs) of SARS-CoV-2 are recognized by endosomal pattern recognition receptors (PRRs), such as Toll-like receptors, which trigger intracellular signaling cascades, leading to the recruitment of neutrophils, activation of macrophages, and transcriptional activation of interferon regulatory factor (IRF) genes (Kawai and Akira, 2011). Furthermore, PAMPs recognition can also activate the NF-κB pathway, which leads to the production and signal transduction of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), driving the inflammatory response (Saha et al., 2010; Hadjadj et al., 2020; Kim et al., 2021).
Interferon (IFN) is an essential component of the innate immune response and plays a crucial role in combating viral infections. Recognition of viral infections by innate immune sensors activates type I and type III IFN response (Park and Iwasaki, 2020). Type I and type III IFNs then induce hundreds of antiviral effectors, or Interferon-Stimulated Genes (ISGs), to establish a cell-intrinsic state of viral resistance (Schneider et al., 2014; Park and Iwasaki, 2020). Several SARS-CoV-2 proteins, such as open reading frame 6 (ORF6) and ORF3b, have the ability to suppress the IFN response (Konno et al., 2020; Park and Iwasaki, 2020; Minkoff and tenOever, 2023). Studies showed that serum IFN activity in severe or critical COVID-19 patients was significantly low, with striking downregulation of ISGs (Blanco-Melo et al., 2020; Hadjadj et al., 2020), indicating an impaired IFN response. When the IFN response is insufficient to control initial viral replication, delayed IFN could lead to inflammation and lung injury (Lee J. S. et al., 2020; Park and Iwasaki, 2020; Domizio et al., 2022; Minkoff and tenOever, 2023). As infected cells die, inflammatory substances associated with the virus are released into the extracellular space, enabling nearby cells to induce an IFN response (Minkoff and tenOever, 2023). The sustained increase in the levels of type I IFNs in the late phase of the infection promotes the accumulation of monocytes-macrophages and the production of pro-inflammatory cytokines, resulting in lethal pneumonia with vascular leakage and impaired virus-specific T cell responses (Lee J. S. et al., 2020). Additionally, this delayed IFN response can lead to systemic inflammation, which is associated with adverse clinical outcomes (Park and Iwasaki, 2020; Domizio et al., 2022; Minkoff and tenOever, 2023).
On the other hand, active virus replication and release cause host cells to undergo pyroptosis, releasing damage-associated molecular patterns (DAMPs) that neighboring epithelial cells, endothelial cells, and alveolar macrophages recognize, initiating the production of pro-inflammatory cytokines and chemokines, which attract monocytes, macrophages, and T cells to the site of infection, promoting further inflammation and establishing a pro-inflammatory feedback loop (Figure 2; Tay et al., 2020). Furthermore, increasing evidence suggests that overactivation of the complement system may be one of the pathogenic mechanisms of SARS-CoV-2 (Holter et al., 2020; Mastellos et al., 2020).
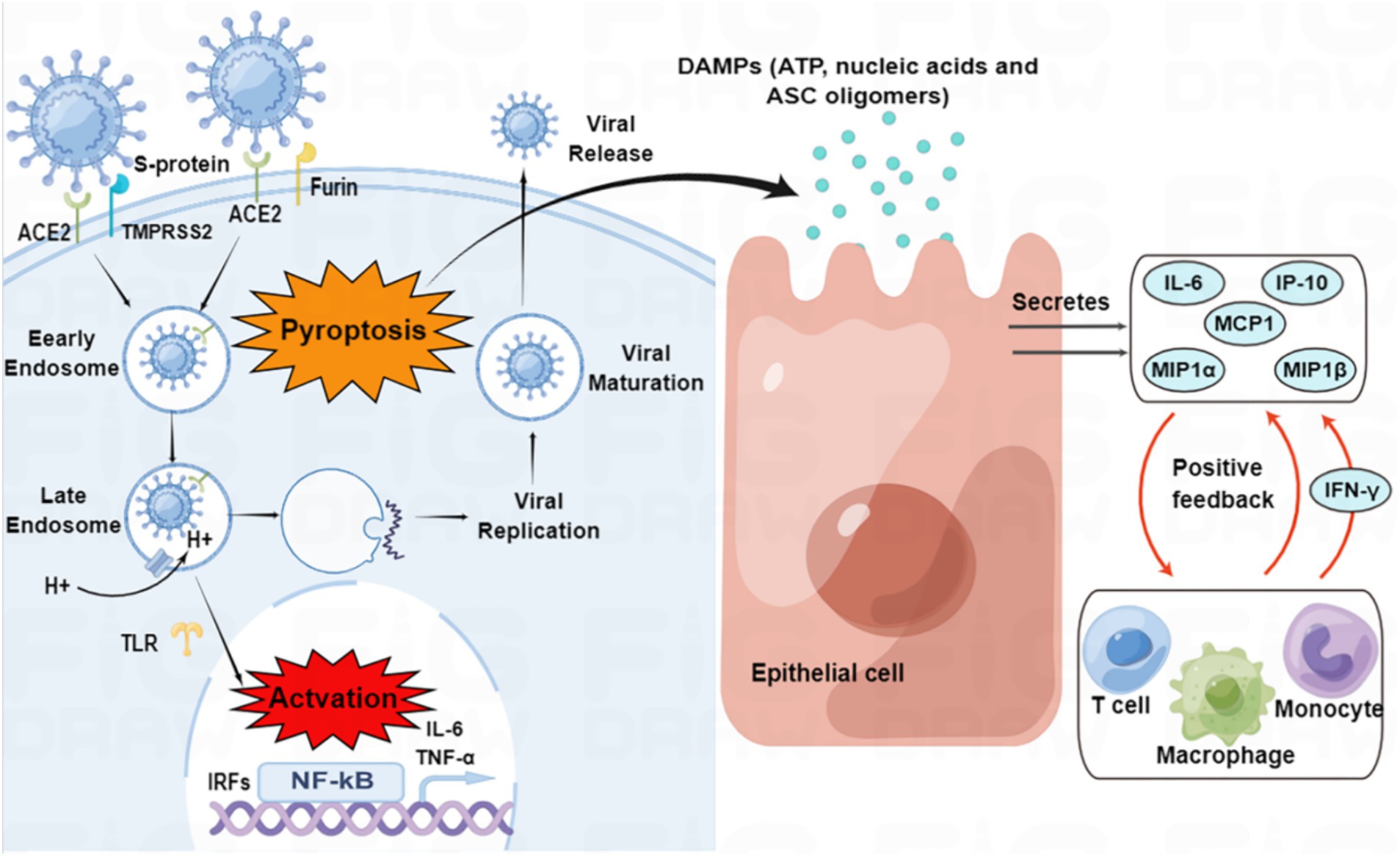
Figure 2. SARS-CoV-2 entry and immune activation. SARS-CoV-2 enters the cell by binding to angiotensin-converting enzyme 2 (ACE2) through transmembrane serine protease 2 (TMPRSS2)-enhanced endocytosis. Furin can also facilitate viral entry. The pathogen-associated molecular patterns (PAMPs) of SARS-CoV-2 are recognized by endosomal pattern recognition receptors, including Toll-like receptors (TLRs), which activate interferon regulatory factor (IRF) genes and nuclear factor kappa B (NF-κB). Pyroptosis of host cells releases damage-associated molecular patterns (DAMPs), which neighboring epithelial cells, endothelial cells, and alveolar macrophages recognize, triggering the production of pro-inflammatory cytokines and chemokines, such as interleukin (IL)-6, interferon γ-induced protein 10 (IP-10), macrophage inflammatory protein 1α (MIP1α), MIP1β and monocyte chemoattractant protein 1 (MCP1). These cytokines and chemokines attract monocytes, macrophages, and T cells to the site of infection, promoting further inflammation and establishing a pro-inflammatory feedback loop.
4.1.2 The role of SARS-CoV-2 accessory proteins in innate immune responseSARS-CoV-2 accessory proteins have the capacity to regulate various aspects of host immunity, including stress response, autophagy, apoptosis, and innate immunity (Zandi et al., 2022). The majority of these accessory proteins act as interferon antagonists, including ORF6, ORF3b, ORF7a, ORF7b, ORF8, ORF9b, and ORF10, impeding the IFN response through diverse mechanisms. For instance, ORF6 prevents the phosphorylation of STAT to inhibit IFN activation, while polyubiquitination of ORF7a can suppress the IFN-I response by inhibiting STAT2 (Zandi et al., 2022). ORF10 has exhibited the ability to suppress the IFN-I signaling pathway by interacting with mitochondrial antiviral signaling protein (MAVS; Li X. et al., 2022; Zandi, 2022). Additionally, ORF3a and ORF3b modify crucial cellular processes such as apoptosis and autophagy (Wong and Perlman, 2022; Zandi et al., 2022). The ectodomain of ORF7 binds to CD14+ monocyte markers, potentially attenuating antigen-presenting cell (APC) function and leading to overexpression of proinflammatory cytokines. This interaction between ORF7a and monocytes suggests its potential role in recruiting monocytes to the lungs during COVID-19 (Zandi et al., 2022). The ORF8 protein downregulates the presentation of viral antigens via the class I major histocompatibility complex (MHC-I), thus dampening the host’s innate immune response (Zandi et al., 2022). Upregulation of ORF9b induces autophagy. ORF9b can also activate inflammasome as a means of evading immune responses (Zandi et al., 2022). Furthermore, the ORF9c accessory protein of SARS-CoV-2 interferes with antigen presentation, interferon signaling, and other immune and stress pathways in human lung epithelial cells (A549 cell line), suggesting its potential involvement in immune evasion processes. However, it should be noted that these experiments were performed using in vitro overexpression systems, and the expression of these proteins in virally infected cells within the context of an in vivo SARS-CoV-2 infection remains unclear (Zandi, 2022). Overexpression of ORF10 triggers the process of mitophagy by promoting the accumulation of LC3 in mitochondria (Li X. et al., 2022; Zandi, 2022). Both ORF3a and ORF7a have been reported to induce the expression of inflammatory cytokines through the activation of NF-κB signaling (Wong and Perlman, 2022).
4.2 Adaptive immune response 4.2.1 Cellular immunitySARS-CoV-2 parasitizes within host cells, relying on them for replication and survival, thus cellular immunity plays a crucial role in eliminating viral infection. However, an abnormal activation and differentiation of T cells occurs in severe cases of SARS-CoV-2 infection (Kalfaoglu et al., 2020). The first autopsy of COVID-19 revealed a substantial reduction in peripheral CD4+ or CD8+ T cell counts, while their status was hyper-activated (Xu Z. et al., 2020). The proportion of highly pro-inflammatory CCR6+ Th17 cells in the CD4+ T cell population was increased (Xu Z. et al., 2020). Patients with severe SARS-CoV-2 infection exhibited a significant decrease in the proportion of multifunctional CD4+ T cells compared to those with mild infection, suggesting that the function of CD4+ T cells had been compromised (Zheng et al., 2020). Furthermore, CD8+ T cells were found to have high concentrations of cytotoxic granules, with 31.6% of the cells being perforin-positive, 64.2% granulysin-positive, and 30.5% double positive for granulysin and perforin (Xu Z. et al., 2020). CD8+ T cells showed high granular enzyme cytotoxicity, with increased expression levels of immune checkpoint inhibitory receptors programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and T cell immunoreceptor with Ig and ITIM domains (TIGIT; Zheng et al., 2020). Following SARS-CoV-2 infection, the levels of CD38+ T cells expressing depletion markers were significantly increased (Wauters et al., 2021). These studies suggest that both elevated exhaustion levels and reduced functional diversity of T cells in severe cases of SARS-CoV-2 infection may be associated with the progression of COVID-19 patients (Zheng et al., 2020). Moreover, T cells in patients with severe COVID-19 express high levels of CD25, which produces the protease furin that facilitates the cell entry of SARS-CoV-2 (Kalfaoglu et al., 2020).
Lymphopenia is one of the main features of severe SARS-CoV-2 infection (Tay et al., 2020; van Eijk et al., 2021). The virus has been shown to induce programmed T cell death, such as apoptosis, contributing to lymphopenia. Reports of reduced peripheral blood T cell levels in patients suggest that T cells are stimulated to exit the blood and migrate to sites of infection to control the viral infection (Tay et al., 2020). van Eijk et al. (2021) proposed that abnormal innate immune responses, direct SARS-CoV-2 infection of T cells, virus-induced tissue damage of lymphatic organs, cytokine-induced apoptosis and pyroptosis of lymphocytes, macrophage activation syndrome (MAS)-associated hemophagocytosis, lymphocyte sequestration in lungs or other organs, and reduced bone marrow hematopoiesis can all contribute to COVID-19-associated lymphopenia. These explanations represent the most comprehensive understanding of lymphopenia associated with COVID-19 to date.
4.2.2 Humoral immunityAntigen-presenting cells (APCs) recognize antigens and stimulate the body’s humoral immunity via virus-specific B and plasma cells to produce neutralizing antibodies. Neutralizing antibodies prevent infection by blocking S protein interaction with ACE2 receptors and viral uncoating (Forchette et al., 2021). Antibody responses to SARS-CoV-2 typically arise within 4–8 days after symptom onset, and most patients seroconvert within the first 3 weeks (Liang et al., 2020; Tay et al., 2020; To KK et al., 2020). The production of IgM is an early and transient response to new antigens, while IgG predominates as a long-term antibody with a longer half-life and lower molecular weight, providing long-term protection and effective tissue penetration (Liang et al., 2020).
Antibodies play a crucial role as a fundamental component of protective immunity against pathogens. However, when an antibody against a pathogen—whether acquired by an earlier infection, vaccination or passive transfer—worsens its virulence through mechanisms dependent on antibodies, the severity of the disease in infected individuals or animals is enhanced. This phenomenon is known as antibody-dependent enhancement (ADE; Arvin et al., 2020). ADE has been documented to occur through two distinct mechanisms in viral infections. Generally, antibodies bind viruses to immune cells via Fcγ receptors on the cell surface and internalization of viruses typically results in their degradation. If antibody binding enhances the ability of viral proteins to enter target cells, or if the virus possesses the capability to evade destruction and produce more viruses after Fcγ receptor-mediated entry, the infection will be amplified. Additionally, if antibodies that bind viruses and Fcγ receptors on cells of the immune system lead to excessive release of cytokines, or if there is an excessive activation of complement due to the abundant formation of immune complexes between antibodies and viral proteins (antigens), it can result in increased disease severity (Arvin et al., 2020; Bournazos et al., 2020; Lee W. S. et al., 2020). ADE of disease is a general concern for the development of vaccines and antibody therapies, and it has been observed in various viruses such as respiratory syncytial virus and dengue virus (Lee W. S. et al., 2020). During the early stages of the COVID-19 pandemic, scientists indeed expressed concern about the potential occurrence of ADE of the disease. However, as research progressed and more data was accumulated, no definitive evidence has been found to support the existence of ADE in COVID-19.
5 Cytokine storm and organ involvement 5.1 Cytokine storm“Cytokine storm” is a condition of uncontrolled systemic hyper-inflammation caused by cytokine excess, which can lead to MOF, and even death (Yang et al., 2020; Kim et al., 2021). SARS-CoV-2 infection causes ACE2 receptor internalization, with a subsequent reduction in the plasma membrane levels of ACE2, resulting in overactivation of the ACE/Ang II/AT1 receptor axis and loss of ACE2/Ang1-7/Mas receptor axis, eventually leading to the overproduction of inflammatory factors (Mahmudpour et al., 2020; Verdecchia et al., 2020). A dysfunctional immune response also contributes to the cytokine storm (Kim et al., 2021). Macrophages are one of the main components of the immune system that are involved in creating the cytokine storm, and these are highly prone to polarization toward the M1 phenotype following SARS-CoV-2 infection, which leads to the release of excessive amounts of inflammatory factors (Nazerian et al., 2021). Aberrant activation of NF-κB caused by SARS-CoV-2 infection also leads to the expression of transcription factors that may drive this exacerbated inflammation (Hadjadj et al., 2020). Furthermore, the complement component C3a promotes differentiation of CD16-expressing highly cytotoxic T cells, which subsequently promotes the release of neutrophils and monocyte chemoattractants (Georg et al., 2022). The cytokine storm arises in part from an abnormal IFN response (Kim et al., 2021), which was supported by a study by Blanco-Melo et al. (2020), who performed transcriptome profiling of respiratory cell types to show that SARS-CoV-2 infection elicits exceptionally low IFN levels while inducing a robust pro-inflammatory cytokine response (Figure 3).
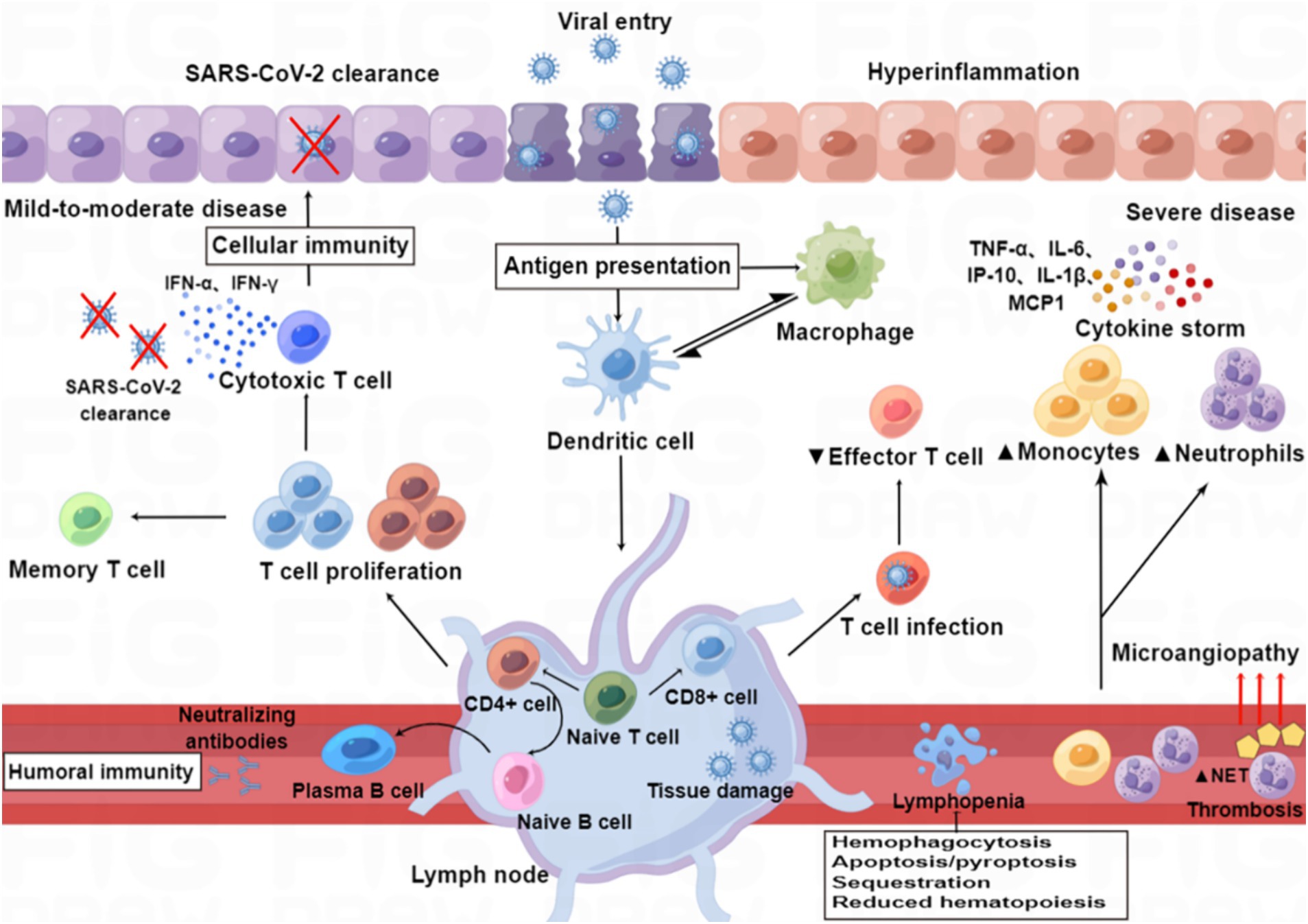
Figure 3. Immunopathology of SARS-CoV-2 infection. Patients with mild to moderate COVID-19 can clear the virus with a healthy immune response, while those with severe infection show immune dysfunction, characterized by relatively increased levels of neutrophils and monocytes and decreased levels of effector T lymphocytes. SARS-CoV-2 infection induces an excess of cytokines, which contribute to hyper-inflammation as constituents of the “cytokine storm” in severe disease (e.g., IL-6, IP-10, IL-1β, TNF-α, and MCP-1), whereas others are particularly important for viral clearance in mild to moderate disease (e.g., IFN-α and IFN-γ). SARS-CoV-2 infection also induces the release of neutrophil extracellular traps (NETs), and excessive formation of NETs leads to a strong pro-coagulant response. Lymphopenia may be caused by hemophagocytosis, reduced hematopoiesis and increased apoptosis/pyroptosis.
Previous studies have shown that various cytokines, including IL-1 family members, IL-6, IL-8, IL-10, TNF-α, IFN-γ, IFN-γ-induced protein 10 (IP-10), monocyte chemoattractant protein 1 (MCP1) and granulocyte colony-stimulating factor (GM-CSF), are elevated in severe SARS-CoV-2 infection (Huang et al., 2020; Mortaz et al., 2020; Kim et al., 2021). These pro-inflammatory cytokines play important roles in the acute phase of inflammation. IL-6 promotes the production of several acute-phase proteins, priming inflammatory responses (McGonagle et al., 2020a). GM-CSF links T-cell-driven acute pulmonary inflammation with an autocrine, self-amplifying cytokine loop, leading to monocyte and macrophage activation (Mortaz et al., 2020).
5.2 Organ involvement 5.2.1 Lung involvementThe abnormal host response or immune system’s overreaction to SARS-CoV-2 can result in the production of extremely high levels of inflammatory cytokines, chemokines and free radicals, causing severe damage to multiple organs (Mortaz et al., 2020). The typical clinical manifestations of lung involvement include fever, dry cough and dyspnea (Bourgonje et al., 2020). Although most patients experience mild symptoms, a significant proportion of patients develop the more serious condition, namely viral pneumonia and ARDS (Huang et al., 2020). Autopsy studies show that the lung pathology is diffuse alveolar damage rather than diffuse alveolar hemorrhage, supporting the role of ARDS in COVID-19 mortality (Liu et al., 2020; Xu Z. et al., 2020). Cytokine storm may be a crucial mechanism underlying ARDS. Elevated levels of IL-1 in SARS-CoV-2 infection lead to the accumulation and activation of a large number of neutrophils in the lung, wherein oxygen free radicals, proteases and inflammatory mediators are released through a “respiratory burst,” causing damage to target cells, which eventually leads to alveolar cell loss, hyaline membrane formation and pulmonary edema, limiting lung gas exchange and causing respiratory distress and hypoxaemia (Tay et al., 2020; Ronit et al., 2021). The levels of C-X-C motif chemokine ligand 10 (CXCL10)/IP-10 and GM-CSF in patients with COVID-19 ARDS were also found to be elevated (Hue et al., 2020), which result in the perpetuation or further amplification of the inflammatory process, respectively. However, the role of cytokine storm in COVID-19-associated ARDS is still under debate since some studies have shown low pro-inflammatory cytokine levels compared to archetypical conditions associated with MAS (Leisman et al., 2020).
Smoking may increase the risk of ARDS due to the upregulation of ACE2 in the airways, which facilitates virus entry (Leung et al., 2020). Obesity may also be a risk factor for ARDS, since visceral fat is able to induce pro-inflammatory effects (Jose and Manuel, 2020). Poor chest wall elasticity and respiratory system compliance in obese patients lead to impaired lung function (Gao et al., 2021). Diabetic patients with elevated levels of IL-6 may have an increased risk of thrombosis that leads to blood-air barrier dysfunction (Gao et al., 2021), thereby increasing the risk of ARDS. Hypertension may also increase the risk of ARDS due to downregulation of ACE2/Ang(1-7) and upregulation of ACE/Ang II, leading to increased inflammatory responses (South et al., 2020). These comorbidities may contribute to the progression of COVID-19 patients to ARDS.
5.2.2 Thromboembolic riskSARS-CoV-2 infection can cause vascular changes such as severe endothelial injury, disrupted endothelial cell membranes, and widespread vascular thrombosis with microangiopathy, ultimately lead to multi-organ failure (Ackermann et al., 2020; Perico et al., 2021). Infection-mediated endothelial injury and endothelialitis, found in multiple vascular beds (including the lungs, kidney, heart, small intestine, and liver) in patients with COVID-19, can trigger excessive thrombin production, inhibit fibrinolysis, and activate complement pathways, initiating thromboinflammation and ultimately leading to microthrombi deposition and microvascular dysfunction (Gupta et al., 2020). However, the finding that viral RNA is rarely detectable in blood indicates that systemic endothelial dysfunction and vasculopathy in COVID-19 patients are not caused by a direct effect of the virus on endothelial cells. The hypercytokinaemia and the massive pro-inflammatory response of the host may contribute to endothelial dysfunction in COVID-19, particularly via the actions of IL-6 and TNF (Teuwen et al., 2020).
Excessive complement activation has been shown to cause intravascular coagulation (Noris et al., 2020). Activation of the complement system leads to endothelial cell injury and death with subsequent vascular denudation and exposure of the thrombogenic basement membrane, which drives activation of clotting cascades (Perico et al., 2021). In addition, the damage and inflammation in endothelial cells lead to the release of PAMPs and DAMPs, which are recognized by monocytes with expression of pattern PRRs, resulting in a high expression of tissue factor in them, thereby activating the extrinsic coagulation pathway (van Eijk et al., 2021). Furthermore, viral infection induces the release of neutrophil extracellular traps (NETs) composed of neutrophil-derived DNA and acetylated histones, which capture and kill invading pathogens as part of the innate immune. However, the excessive formation of NETs can cause a strong pro-coagulant response and are found in various organs of COVID-19 patients (Brinkmann et al., 2004; Middleton et al., 2020; Schurink et al., 2020; Borczuk, 2021). The presence of high levels of platelet factor 4 (PF4), IL-6 and IL-8 in the serum can induce the formation of NETs (Rossaint et al., 2014; Middleton et al., 2020). An increased formation of NETs was a potential biomarker of disease severity. Elevated D-dimer levels reflect vascular bed thrombosis with fibrinolysis (Tang et al., 2020a,b; McGonagle et al., 2020b). Antiphospholipid antibody levels are elevated in critical COVID-19 patients, increasing the risk of (micro)vascular thrombosis (MIYAKIS et al., 2006; Zhang Y. et al., 2020).
5.2.3 Neurological involvementSome COVID-19 patients may experience neurological symptoms such as headache, confusion, anosmia, taste disturbance, nausea, and vomiting (Mao et al., 2020). Anosmia is the most common and specific neurological presentation in COVID-19 patients. The infection of non-neuronal sustentacular cells in the olfactory mucosa by the SARS-CoV-2 and the subsequent local inflammation are associated with the anosmia (Minkoff and tenOever, 2023). Persistent anosmia in certain COVID-19 patients may be due to axon injuries and microvasculopathy in olfactory tissue, as suggested by higher mean axon pathology scores and microvasculopathy scores, lower axon density, and ultrastructural changes in olfactory axons (Ho et al., 2022). In addition, SARS-CoV-2 infection causes widespread downregulation of olfactory receptors (ORs) and of their signaling components (Zazhytska et al., 2022). However, whether olfactory neurons can be infected by SARS-CoV-2 remains controversial (de Melo et al., 2021; Khan et al., 2021).
Central nervous system (CNS) manifestations are the major forms of neurological involvement seen in COVID-19 patients (Mao et al., 2020). Systemic inflammation-induced neuroinflammation, consequent glial dysregulation and impairment of neural function represents a pathological mechanism of COVID-19 (Fernández-Castañeda et al., 2022). Autopsy findings showed hyperaemic and edematous brain tissue, neuronal degeneration, and activation of microglia with the formation of nodules (Mao et al., 2020; Schurink et al., 2020). Following mild respiratory COVID in mice, persistently impaired hippocampal neurogenesis, decreased oligodendrocytes, and myelin loss were evident together with elevated cerebrospinal fluid (CSF) cytokines/chemokines including CCL11. Concordantly, humans who experience persistent cognitive symptoms after COVID-19 exhibit elevated CCL11 levels (Fernández-Castañeda et al., 2022). In the brains of deceased COVID-19 patients, extensive inflammation was seen in the olfactory bulbs and medulla oblongata (Schurink et al., 2020). Furthermore, in the context of pro-inflammatory hypercoagulable state of COVID-19, acute cerebrovascular disease is also emerging as an important complication (Ellul et al., 2020). Viral RNA has been detected in the CSF and other brain tissues collected from patients who died from COVID-19 (Ellul et al., 2020; Matschke et al., 2020). However, the direct role of the virus in the neurological manifestations and the portal of entry of SARS-CoV-2 in the CNS remain highly debated (de Melo et al., 2021).
A novel neurological manifestation called COVID-19 hemiencephalitis has recently been observed. This condition is characterized by hyperintensity in one cerebral hemisphere on fluid-attenuated inversion recovery images and subsequent brain atrophy (Wang and Yin, 2024). Additionally, a study found an increased incidence of functional neurological disorders (FNDs) associated with long-COVID (Alonso-Canovas et al., 2023). Reports of neurological sequelae, commonly referred to as “brain fog,” are also becoming more prevalent. According to Greene et al., persistent localized blood–brain barrier (BBB) dysfunction and sustained systemic inflammation are key features of long COVID-related brain fog. These findings suggest that targeted regulation of BBB integrity may offer a new approach for managing patients with long-COVID (Greene et al., 2024).
6 Evolution and mutationThe Wuhan-Hu-1 is not the direct ancestor of all early new coronaviruses worldwide. It has resulted from three α mutations, consisting of two synonymous and one non-synonymous mutation in the ancestral strain (Kumar et al., 2021). However, the exact origin has not yet been determined. Studies indicate that SARS-CoV-2 may have entered the human population earlier than 2019 through unnoticed infections. During this period of cryptic transmission, the virus could have gradually acquired key mutations, including the RBD and furin cleavage site insertions, which facilitated its adaptation to humans (Zhang and Holmes, 2020). The acquisition of the furin cleavage site by SARS-CoV-2 and subsequent cleavage of the S protein is essential for human infection. The virus retained this cleavage site throughout the pandemic (Jackson et al., 2022).
SARS-CoV-2 has the ability to produce significant genomic changes (Davies et al., 2021; Harvey et al., 2021). In general, as viruses evolve to escape immune surveillance, they often undergo reduced fitness and become less infectious (Dazert et al., 2009; das et al., 2011; Sui et al., 2014; Shang et al., 2020). However, SARS-CoV-2 remains highly infectious despite being immune evasive, which may contribute to its widespread transmission (Chu et al., 2020; Sanche et al., 2020; Shang et al., 2020).
6.1 Key mutations and variantsDuring the early stages of the pandemic, SARS-CoV-2 S protein contained the aspartic acid (D) at position 614. As the pandemic progressed, however, a variant containing glycine at position 614 emerged and rapidly became dominant (Butowt et al., 2020; Boehm et al., 2021). One possible reason for the emergence of this mutation could be the furin cleavage site in the S protein of SARS-CoV-2 makes it more prone to S1 shedding, which reduces its infectivity compared to SARS-CoV. To compensate for this disadvantage, the SARS-CoV-2 S protein has seemingly developed a stronger intermolecular association between the S1 and S2 subunits through the D614G mutation (Jackson et al., 2022). Furthermore, the D614G mutation favors the open conformational state of the S protein, leading to increased RBD exposure and higher binding affinity to the ACE2 host receptor (Butowt et al., 2020). Regardless of its mechanism for enhanced infectivity, the widespread presence of the D614G mutation in most subsequent SARS-CoV-2 isolate suggests that it is indeed a beneficial mutation for adaptation to humans.
In addition to the D614G mutation, the SARS-CoV-2 variants Alpha (lineage B.1.1.7), Beta (B.1.351) and Gamma (P.1), which were first identified in the United Kingdom, South Africa and Brazil, respectively, carry a common N501Y mutation (Andreano et al., 2021; Faria et al., 2021; Harvey et al., 2021; The COVID-19 Genomics UK (COG-UK) consortium et al., 2021; Tian et al., 2022). The N501Y mutation substitutes asparagine with tyrosine at position 501, forms a potential aromatic ring-ring interaction and an additional hydrogen bond with ACE2, and reduces the polarity of critical residues in RBD, thereby increasing the affinity between RBD and the cellular surface of ACE2 (Tian et al., 2022). The Delta (B.1.617.2), first identified in India, does not carry the N501Y mutation. However, it possesses the P681R mutation in the furin cleavage site, which might contribute to increased transmissibility of the variant. Additionally, the Delta variant (B.1.617.2) also carries the L452R mutation, which causes moderate loss of susceptibility to neutralizing antibodies, allowing the variant to escape the human HLA-A24-presented T-cell response (Motozono et al., 2021). Many studies showed Delta (B.1.617.2) was associated with increased severity of illness and risk of death (Fisman and Tuite, 2021; Ong et al., 2022). There has been controversy regarding whether the Delta (B.1.617.2) variant exhibits higher viral loads in the upper respiratory tract. Some studies have suggested increased viral loads associated with the Delta variant (Costa et al., 2022; Li B. et al., 2022). However, in a cohort study conducted by Ong et al. (2022), they reported comparable SARS-CoV-2 Delta and Alpha RNA loads in nasopharyngeal specimens (NP) at the time of SARS-CoV-2 infection diagnosis using RT-PCR. Further research is needed to better understand the viral load dynamics and transmissibility of the Delta variant.
The COVID-19 pandemic enters a longer phase where immune escape will play a significant role in shaping the evolution of the S protein (Jackson et al., 2022). As the virus continues to circulate and encounters immune responses from vaccinated or previously infected individuals, selective pressure may drive the emergence of new variants with mutations that allow them to evade the immune system. E484K has been identified as an escape mutation that emerges during exposure to monoclonal antibodies C121 and C144 and convalescent plasma, and was the mutation described as able to reduce the neutralizing ability of a combination of monoclonal antibodies (REGN10989 and REGN10934) to an unmeasurable level in convalescent (Baum et al., 2020; Weisblum et al., 2020). The E484K mutation reduces binding of neutralizing antibodies and leads to partial immune escape, decreasing the efficacy of some antibody therapies or vaccines in vitro (Boehm et al., 2021). The combination of K417T + E484K + N501Y mutations may be more effective than any of these mutations alone in decreasing the neutralization of antibodies (Greaney et al., 2021). The SARS-CoV-2 variants Beta (B.1.351), Gamma (P.1) and Delta (B.1.617.2) have all experienced mutations at the E484 position.
The Omicron (B.1.1.529) variant, first identified in South Africa and Botswana, has attracted worldwide attention since its emergence owing to its high transmissibility and immune evasion capability (Fan et al., 2022; Shrestha et al., 2022). The Omicron variant did not develop from one of the earlier known variants, the phylogenetic analysis revealed that the Omicron variant formed a new monophyletic clade that is distant from other SARS-CoV-2 variants (Kandeel et al., 2022). There are three possible explanations for the development of the Omicron variant: silent evolution in a population with little sequencing, long-term evolution in one or a few individuals with chronic infection, or evolution in other animals (Fan et al., 2022; Shrestha et al., 2022). Some studies have suggested that the pre-outbreak mutations in Omicron might have accumulated in a non-human host, particularly rodents (Wei et al., 2021; Mallapaty, 2022; Smyth et al., 2022). The Omicron variant is not a single strain, but comprises three distinct sub-lineages: BA.1, BA.2, and BA.3. Subsequently, two other sub-lineages have also been identified, BA.4, BA.5 (Shrestha et al., 2022). The Omicron variant carries the highest number of mutations. In the BA.1 lineage, up to 60 mutations have been identified. Among these, as many as 38 mutations occur in the S protein, one in the envelope E protein, two in the membrane M protein, and six in the N protein. BA.2 lineage possesses 57 mutations, with 31 in the S protein (Fan et al., 2022; Kannan et al., 2022). Laboratory studies showed that the mutations in the Omicron variant altered the tropism of the virus. The Omicron tended to infect the upper respiratory tract, which can be attributed to that Omicron inefficiently utilize TMPRSS2 to enter cells, but mainly rely on the endocytic pathway, leading to a decrease in replication in the lung parenchyma and an enhanced ability to infect the upper respiratory tract (Hui et al., 2022; Meng et al., 2022). Increasing studies have indicated that infection with Omicron is associated with milder symptoms compared to earlier variants (Abdullah et al., 2022; Ulloa et al., 2022; Wolter et al., 2022). The Omicron variant exhibits a reduced capacity to induce syncytia formation in tissue culture. Syncytia formation has been associated with increased disease severity (Meng et al., 2022; Suzuki et al., 2022). However, all these data were obtained in in vitro or animal models, while it is likely that the reduced pathogenicity of the Omicron variant is due to the rising levels of cell-mediated immunity resulting from previous infections and vaccinations (Fan et al., 2022). The Omicron variant is capable of evading the humoral immune response established by vaccination or previous infection with other variants. Additionally, most therapeutic monoclonal antibodies are ineffective against the Omicron variant. These are attributed to multiple mutations and deletions in the S protein of the Omicron variant render a part of the S protein unrecognizable to the antibodies developed by natural infection or vaccination (Zhang and Holmes, 2020; Zhang et al., 2021; Iketani et al., 2022; VanBlargan et al., 2022). Fortunately, components of the cellular immune response, such as T cells, are still able to target Omicron and provide protection from severe outcomes. Data from La Jolla Institute has revealed that, despite several mutations in S protein, on average 94% of CD8 and 91% of CD4 epitopes of Omicron are still completely conserved, suggesting this variant has not evolved extensive T-cell escape mutations at this time (Shrestha et al., 2022).
The SARS-CoV-2 Omicron sub-variant BA.2.86 was first identified in Denmark in August 2023 (Rasmussen et al., 2023). Compared to its ancestor BA.2, the BA.2.86 sub-variant of Omicron carries 29 substitutions, 4 deletions, and 1 insertion in the S protein, suggesting an increased ability to evade antibodies induced by vaccination and previous infections (Looi, 2023; Mahase, 2023). The emergence of BA.2.86 highlights the continuous evolution of the SARS-CoV-2, influenced by global immunity levels, vaccination campaigns, and treatment strategies (Satapathy et al., 2024).
6.2 QuasispeciesConsensus genomic sequences of SARS-CoV-2 variants represent the genomes most frequently found in the clinical samples of patients, which have been widely used to monitor the global spread of the virus during the COVID-19 pandemic (Colson et al., 2023; Messali et al., 2023). However, these genomes represent the most frequently observed viral genomes and do not capture the full extent of their diversity (Colson et al., 2023). With advancements in sequencing techniques, studies have reported intrahost genetic diversity of SARS-CoV-2 in specific patient groups such as immunocompromised individuals, chronically infected patients, those treated with monoclonal antibodies targeting the spike protein, and within different body compartments of a single patient (Colson et al., 2023). This suggests that SARS-CoV-2 infections consist of a population of closely related viral variants, referred to as quasispecies (Colson et al., 2023; Messali et al., 2023).
A viral quasispecies is defined as the dynamic distribution of nonidentical but closely related mutants, variants, recombinant, or reassortant viral genomes (Colson et al., 2023; Messali et al., 2023; Delgado et al., 2024). This concept reflects the understanding that populations typically consist of distinct “types” characterized by certain criteria rather than fully identical individuals. Consequently, virus isolates or strains cannot be considered as linked to a single sequence but rather encompass a collection of sequences (Colson et al., 2023). It is currently hypothesized that hosts are infected by a diverse ensemble of viruses, termed the “wild bunch,” rather than multiple virions sharing the same genome. Each virion within this “wild bunch” may exhibit different virulence and tissue tropisms, enabling adaptation to various organs and potentially resulting in diseases of differing nature (Colson et al., 2023). The wide range of symptoms observed in different organs during the acute phase of SARS-CoV-2 infection aligns with the concept of the “wild bunch.” The presence of intrahost genomic variability leads to antigenic diversity and specific phenotypic characteristics, which influence replicative capacity, transmissibility, host range and tropism, susceptibility to immune responses, vaccines, and antivirals (Colson et al., 2023).
Quasispecies has revolutionized our understanding of viral evolution, particularly for RNA viruses. The concept highlights the dynamic and ever-changing nature of viral populations, leading to new insights in viral pathogenesis, vaccine development, and antiviral therapy.
7 TreatmentCurrent therapeutic strategies for SARS-CoV-2 infection include symptomatic treatment, supportive care and repurposed drugs. The main goal of these interventions is to inhibit the entry of the virus into cells. Extrinsic import of human recombinant soluble ACE2 (hrsACE2) can neutralize the virus in the serum (Zhang H. et al., 2020; Nazerian et al., 2021), and antibodies or small molecule drugs with higher binding affinity for the RBD of the virus may block viral attachment (Shang et al., 2020). However, the RBD is poorly conserved across the coronavirus family. Targeting membrane fusion, one of the most conserved regions of the S protein, has potential in treating broad-spectrum pan-CoV disease in the future (Xia et al., 2019; Walls et al., 2020). Xia et al. (2019) identified peptides that target other human coronaviruses with fusion inhibitory activity against membrane fusion domains, suggesting that the S2 protein subunit of SARS-CoV-2 could be a therapeutic target. Aptamers, consisting of single-stranded DNA or RNA molecules, possess the ability to bind to specific targets by adopting a unique 3D structure (Zhang et al., 2023). Currently, there is ongoing development of aptamers targeting the S protein of SARS-CoV-2. Studies have identified MSA52 as a universal aptamer capable of binding to the trimeric S proteins found in various SARS-CoV-2 variants of concern (Zhang et al., 2023). This broad-spectrum binding capability is highly significant for the development of effective antiviral treatments against the virus. As SARS-CoV-2 entry requires endosome acidification, neutralizing the acidic environment of endosomes may impair viral entry pathways (Wang et al., 2020; Nazerian et al., 2021). The antiviral peptide 8P9R was found to be able to cross-link viruses and inhibit endosomal acidification simultaneously to block viral entry (Zhao et al., 2021). Alternatively, TMPRSS2 and lysosomal cathepsins could be targets for preventing viral entry into cells (Hoffmann et al., 2020).
Conventional antivirals such as remdesivir, which inhibit viral RNA synthesis, have in vitro activity against SARS-CoV-2 (Wang et al., 2020). However, remdesivir needs to be administered intravenously, limiting its widespread use during the pandemic. The oral analog of remdesivir, VV116, has improved in vitro antiviral activity and selectivity with satisfactory safety, tolerability, and pharmacokinetic profiles in healthy subjects (Qian et al., 2022). A real-world study demonstrated that VV116 treatment resulted in a marked acceleration of viral shedding in patients infected with the SARS-CoV-2 omicron variant who received VV116 within 5 days after the first positive test (Shen et al., 2022). Compared to paxlovid, a new oral drug consisting of the second-generation protease inhibitor nirmatrelvir co-packaged with the pharmaceutical enhancer ritonavir that has been recommended for use in WHO guidelines, VV116 exhibited a similar time to sustained clinical recovery with fewer safety concerns in symptomatic adults with mild to moderate COVID-19 at risk of progression (Reina and Iglesias, 2022; Cao et al., 2023). Molnupiravir, an oral nucleoside analog with broad-spectrum antiviral activity and resilience to drug resistance, is an isopropyl ester prodrug of β-D-N4-hydroxycytidine (NHC; EIDD-1931) that targets the RdRp (Hadj Hassine et al., 2022). Molnupiravir exerts its antiviral effects through lethal mutagenesis, which increases G → A and C → U transition frequencies during replication to extinguish the virus (Kabinger et al., 2021; Hadj Hassine et al., 2022). It has shown promising efficacy and safety in Phase I/II/III clinical trials, but further studies are needed to evaluate its carcinogenic risks and genotoxicity (Hadj Hassine et al., 2022; Wen et al., 2022). Nanomaterials can be designed and manipulated to selectively target specific cells, reducing drug toxicity (Ao et al., 2023).
There is not enough evidence to support the use of corticosteroids in COVID-19 treatment. If glucocorticoids are used, they should not compromise the host defense mechanisms necessary to resist SARS-CoV-2 infection and must be used in combination with antiviral drugs (Bourgonje et al., 2020; van Eijk et al., 2021). Cytokine receptor antagonists such as anakinra and tocilizumab have been shown to reduce the inflammatory response (Cavalli and Dinarello, 2018; Berardicurti et al., 2020; Somers et al., 2020; Kim et al., 2021), while therapeutic plasma exchange (TPE) rapidly and non-selectively removes excess cytokines from the plasma, altering the proliferation status and function of lymphocytes and enhancing immunity against SARS-CoV-2 (Reeves and Winters, 2014; Knaup et al., 2018; Kim et al., 2021).
Anticoagulant therapy, specifically low molecular weight heparin (LMWH), has been proposed by the International Society of Thrombosis and Hemostasis (ISTH) for use in all hospitalized COVID-19 patients without contraindications (Bourgonje et al., 2020). However, some patients on prophylactic LMWH remain at risk of thrombotic complications due to potential heparin resistance caused by increased levels of NETs, therefore, higher therapeutic doses of LMWH would be required (Middleton et al., 2020). Inhibiting or catabolizing NETs may be a therapeutic avenue to explore, with neonata
留言 (0)