
記住我










Gliomas represent a broad subset of neoplastic diseases within the central nervous system (CNS). Their presentations and prognoses vary based on factors such as precursor cell type, anatomical location, and molecular characteristics (1). Low-grade gliomas have longer survival periods (2) compared with diffuse midline gliomas (DMGs), including diffuse intrinsic pontine gliomas (DIPGs), which are expansile pontine tumors that are usually present in pediatric populations (1, 3). As such, the management principles for glioma widely vary depending on the characteristics of the tumor, ranging from surveillance for low-grade disease progression to palliation for highly aggressive manifestations.
While the diagnosis and classification of gliomas had previously been performed based on anatomic locale, clinical features, and histopathology, the 2021 revision of the World Health Organization's (WHO) classification for CNS tumors has significantly featured molecular characteristics as a means of definitive diagnosis (1). The increasing prevalence of molecular diagnostic techniques in the clinical setting, coupled with targeted therapeutics, makes molecular diagnosis a powerful tool for identifying the most effective treatments for a given tumor. For example, DIPG is now considered a subset of DMG, with H3K27 alteration, characterized by the nominal histone mutation that portends a poor prognosis (1). The management of DIPG typically entails radiation therapy to the pons as its diffuse and aggressive nature precludes surgical resection. Therefore, treatment often proceeds based on characteristic imaging features alone. Due to the risk associated with performing a biopsy on the eloquent tissue of the brainstem, biopsies have traditionally been avoided, although they are being utilized more frequently as researchers attempt to better understand the disease and develop effective therapies for it (4).
This study presents the case of a 19-year-old female who presented with migraine headache, gait instability, and lower extremity paresthesia with brain imaging suggestive of DIPG. However, the tissue diagnosis revealed bland histopathological features associated with a single mutation in the NF1 gene. These findings crucially altered the treatment paradigm following the biopsy.
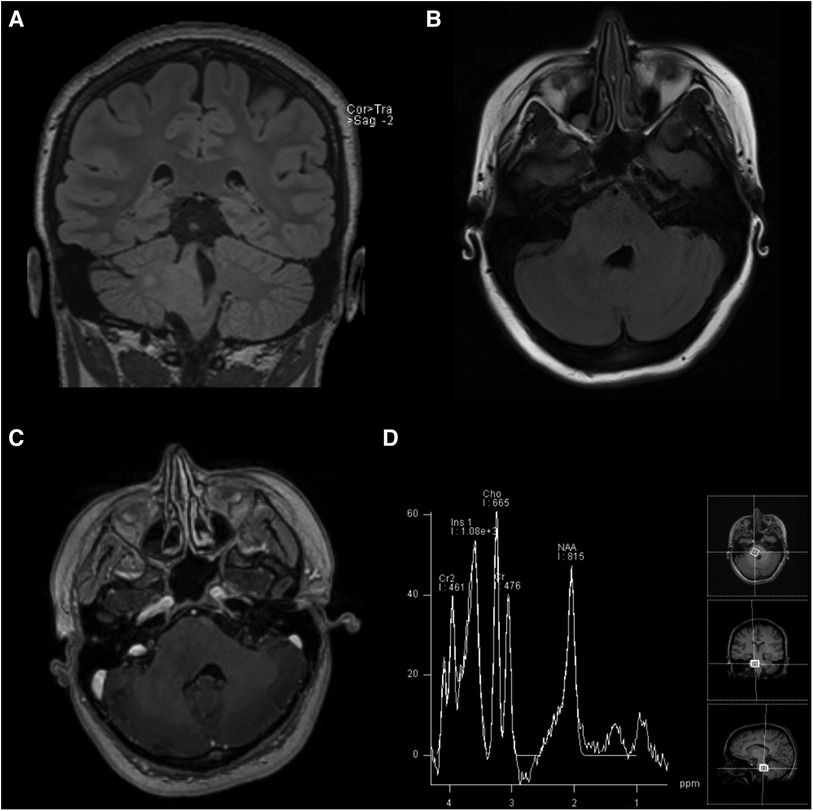
2 Case descriptionA 19-year-old female with a multi-year history of ocular migraines presented to the emergency department with ataxia and lower extremity paresthesia preceded by refractory holocranial migraine. The patient noted that the headaches differed from her previous migraines and had been increasing in severity and duration over the preceding 8 months, culminating in new-onset dizziness, gait instability, and numbness and tingling in her legs over the preceding weeks (Table 1). She was scheduled for neuroimaging because of the progression of symptoms. Magnetic resonance imaging (MRI) revealed an ill-defined mass within the right pons that extended into the right middle cerebellar peduncle and the right posterolateral medulla with a corresponding partial effacement of the fourth ventricle suggestive of DIPG (Figure 1). The supratentorial gray and white matter volumes were within the normal volumetric percentile ranges for her age (5), and subsequent MRI spectroscopy revealed a markedly elevated choline level with decreased NAA in the lesion when compared against the left pons and the middle cerebellar peduncle. In another hospital, the biopsy was initially deferred due to the location of the lesion in the brainstem and the presumptive diagnosis of DIPG, with a recommendation to proceed with standard radiotherapy. However, the patient was transferred to the Brigham and Women's Hospital/Dana-Farber Cancer Institute (DFCI) for further care, where the recommendation to perform a biopsy prior to radiotherapy was made to definitively ascertain the histopathologic and molecular genetic features to determine potential eligibility for ongoing clinical trials.
Table 1. Clinical timeline.
Figure 1. MRI scans taken on initial admission: (A) coronal T2-weighted FLAIR, (B) axial T2-weighted FLAIR, (C) axial post-gadolinium contrast T1-weighted, and (D) magnetic resonance spectroscopy sequences demonstrating the non-enhancing, expansile right pontine lesion with an increased choline peak suggestive of infiltrating glioma.
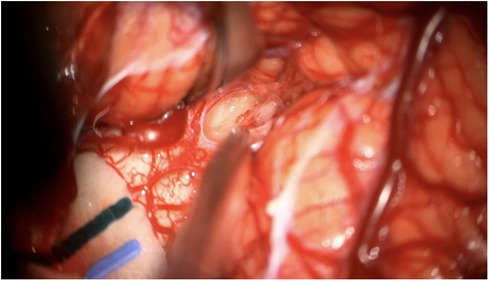
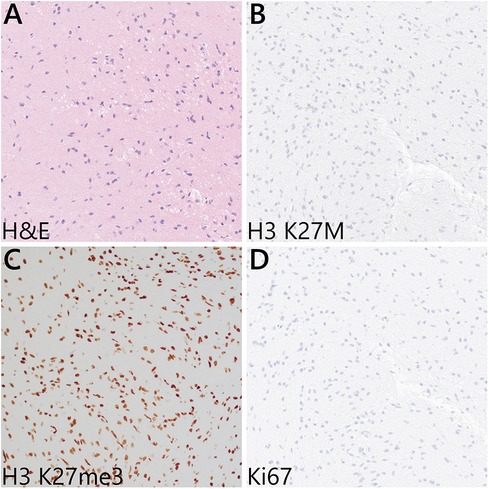
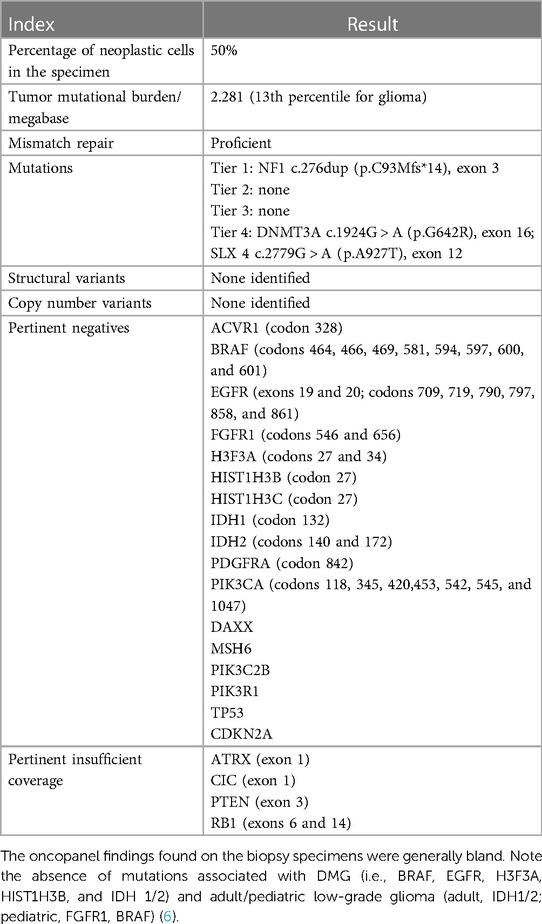
3 Diagnostic assessmentAn open biopsy utilizing neuronavigation and intraoperative electrophysiology was decided upon due to the exophytic components of the mass. A posterior approach with craniotomy to widen the foramen magnum and C1 laminectomy was performed, followed by microsurgical dissection of the right tonsil. Regions of discolored tissue were observed on the floor of the fourth ventricle (Figure 2). A monopolar stimulator was used to map adjacent critical neuroanatomy, and micropituitary cup forceps were used to sample regions of abnormal-appearing tissue, which were silent upon stimulation. The intraoperative frozen section analysis was consistent with the glioma, showing minimal hypercellularity and rare atypical cells. Immunohistochemistry with mutation-specific antibodies against IDH1 R132H and H3K27M was negative, while H3 K27 trimethylation expression was retained (Figure 3). The MIB-1 proliferation index was 0%. Notably, targeted next-generation sequencing (Oncopanel, Table 2) revealed a frameshift variant in exon 3 of the NF1 gene (c.276dup, p.C93Mfs*14) in 36% of the reads with an accompanying loss of 17q heterozygosity. There were no copy number variants, structural variants, or known pathogenic variants in other assayed genes, including IDH1, IDH2, H3F3A, EGFR, FGFR1, PDGFRA, CDKN2A/B, and BRAF, by sequencing. The lack of mutations associated with DMG, namely, H3 K27M/I or alterations in TP53, ACVR1, PDGFRA, or EGFR, retained H3 K27 trimethylation. Low-grade infiltrative histology did not cleanly fit within a current diagnostic entity per the 2021 WHO criteria (1). As a result, the patient was diagnosed with a WHO grade 1 or 2 diffuse low-grade glioma, not otherwise specified.
Figure 2. Intraoperative photograph of the mass lesion taken during the biopsy.
Figure 3. Representative microscopic features from the intraoperative biopsy sample. (A) H&E sections showed mildly hypercellular white matter with infiltrating atypical glial cells—notably, Rosenthal fibers, eosinophilic granular bodies, microvascular proliferation, and necrosis are absent. (B) Immunohistochemistry for H3 K27M was negative, and (C) the H3 K27 trimethylation (me3) expression was retained in the tumor cells. (D) Ki67 labeling was negative.
Table 2. Oncopanel findings.
On postoperative follow-up, the patient recovered well with gradual improvement in her neurologic status and no new neurologic symptoms. She tapered off dexamethasone. The subsequent work-up for neurofibromatosis indicated that the mutation was somatic. She lacked any stigmata of neurofibromatosis upon examination, and the sequencing of the NF1 gene from a peripheral blood sample did not reveal any variants. The follow-up MRI at 3 and 5 months post-biopsy demonstrated a stable lesion. Our patient continues to do well clinically with gradual neurologic improvement. She is participating in physical therapy and has resumed her college classes. Her presenting symptoms of headache, ataxia, and lower extremity paresthesia have remained stable.
4 DiscussionThis study's presentation of low-grade glioma in a region typically associated with DIPG highlights the limitations of neuroimaging in differentiating infiltrative tumors of the CNS and suggests a critical role for molecular diagnosis in such circumstances. In the case of our patient, the mass lesion demonstrated radiographical features consistent with DIPG, particularly effacement of the fourth ventricle, also known as the “flat floor of fourth ventricle sign,” and minimal enhancement on contrast MRI. Although DIPG is the most common form of pontine glioma, the fact that radiographic features are non-specific for DIPG compared to other histopathologic types suggests that imaging characteristics alone may be insufficient for diagnosis (7). The morbidity and mortality associated with brainstem biopsies have been the source of historical apprehension: the most commonly reported complications include cranial nerve palsy (4.2%, in particular, CN VII), perioperative hemorrhage (3.6%), hemiparesis (2.1%), and disorders of speech or movement (2.1 and 1.0%, respectively) (8). However, recent studies suggest that biopsy can safely be done in experienced centers. A meta-analysis by Kickingereder et al. (9), which included 1,480 cases of stereotactic biopsy for brainstem tumors, reported a 96.2% diagnostic success rate at the cost of 1.7% permanent morbidity and 0.9% mortality. A more recent study by Hamisch et al. (10) on 735 cases in the pediatric population reported a 96.1% diagnostic success rate with 0.6% permanent morbidity and 0.6% mortality. The risk of complications secondary to non-indicated therapeutic irradiation must also be considered. In this case, the decision to perform an open biopsy vs. a more common needle biopsy was informed by the dorsally exophitic nature of the lesion, which allowed a direct approach to the tumor with minimal-to-no violation of the adjacent brain.
The importance of molecular diagnosis is further accentuated by the rarity of the genetic profile observed in our patient's tumor. The presence of a putative loss-of-function mutation in the NF1 gene in a subset of reads and in the absence of any of the clinical features associated with neurofibromatosis type 1 (NF1, also known as von Recklinghausen's disease) suggests NF1 mosaicism, a rare phenomenon previously reported in patients with a higher degree of NF1 mutagenic burden in the brain or glioma tissue relative to the somatic tissue (11). The patient did not demonstrate any symptoms of germline NF1 mutation, and the sequencing of a peripheral blood sample did not demonstrate the loss-of-function mutation. There was no family history of neurofibromatosis. In addition, the patient's tumor lacked the genetic markers that are commonly associated with pediatric or adult low-grade glioma, such as mutations in IDH1/2, FGFR1, BRAF, and histone H3 encoding genes (6). We therefore considered the tumor to be related to somatic mosaicism or somatic mutation leading to a biallelic NF1 loss of function. Given the low-grade histologic features, low MIB-1 index, and absence of H3 K27M, we concluded that this tumor is most consistent with low-grade gliomas commonly observed in NF1 patients. Fortunately, the prognosis for these patients is much better than that associated with DIPG. The aggregate findings in our case were consistent with an NF-1 mutant, pediatric low-grade glioma; hence, the decision was made to forego radiation therapy until there is clinical or radiographic evidence of tumor growth during surveillance follow-up.
The generalizability of our findings to other cases of suspected DIPG is limited due to the fact that this is a report of a single case with limited follow-up thus far. However, this case is emblematic because of the marked impact the tissue diagnosis has had on this patient's prognosis and management. Notably, other recent studies have also demonstrated that molecular information obtained from biopsies can guide potential treatment options, including the use of targeted inhibitors (12–15). Nonetheless, given the potential for complications and the incredible heterogeneity in the presentation of pontine glioneuronal masses, we suggest that the decision on whether to perform a biopsy should be based on the clinical judgment of risk and benefit by the surgeon and neuro-oncologist as well as shared decision-making with the patient.
In conclusion, the case presented in this study provides further support to the importance of tumor diagnosis through biopsy and detailed histopathologic and molecular genetic profiling of diffuse brainstem tumors.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author/s.
Ethics statementWritten informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsJB: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. PK: Writing – original draft, Writing – review & editing. JC: Writing – review & editing. JS: Writing – review & editing. GF: Writing – review & editing. DM: Writing – review & editing. KL: Writing – review & editing. DH: Writing – review & editing. DR: Writing – review & editing. PP: Conceptualization, Investigation, Supervision, Writing – review & editing.
FundingThe authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestJB has an equity position in Treovir Inc. and UpFront Diagnostics. JB is also on the Centile Bioscience, QV Bioelectronics, and NeuroX1 boards of scientific advisors.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
PubMed Abstract | Crossref Full Text | Google Scholar
2. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. (2005) 64(6):479–89. doi: 10.1093/jnen/64.6.479
PubMed Abstract | Crossref Full Text | Google Scholar
3. Leach JL, Roebker J, Schafer A, Baugh J, Chaney B, Fuller C, et al. MR Imaging features of diffuse intrinsic pontine glioma and relationship to overall survival: report from the International DIPG Registry. Neuro Oncol. (2020) 22(11):1647–57. doi: 10.1093/neuonc/noaa140
PubMed Abstract | Crossref Full Text | Google Scholar
4. Tejada S, Aquilina K, Goodden J, Pettorini B, Mallucci C, van Veelen ML, et al. Biopsy in diffuse pontine gliomas: expert neurosurgeon opinion-a survey from the SIOPE brain tumor group. Childs Nerv Syst. (2020) 36(4):705–11. doi: 10.1007/s00381-020-04523-8
PubMed Abstract | Crossref Full Text | Google Scholar
5. Bethlehem RAI, Seidlitz J, White SR, Vogel JW, Anderson KM, Adamson C, et al. Brain charts for the human lifespan. Nature. (2022) 604(7906):525–33. doi: 10.1038/s41586-022-04554-y
PubMed Abstract | Crossref Full Text | Google Scholar
8. Dalmage M, LoPresti MA, Sarkar P, Ranganathan S, Abdelmageed S, Pagadala M, et al. Survival and neurological outcomes after stereotactic biopsy of diffuse intrinsic pontine glioma: a systematic review. J Neurosurg Pediatr. (2023) 32(6):665–72. doi: 10.3171/2023.7.PEDS22462
PubMed Abstract | Crossref Full Text | Google Scholar
9. Kickingereder P, Willeit P, Simon T, Ruge MI. Diagnostic value and safety of stereotactic biopsy for brainstem tumors: a systematic review and meta-analysis of 1480 cases. Neurosurgery. (2013) 72(6):873–81. discussion 882; quiz 882. doi: 10.1227/NEU.0b013e31828bf445
PubMed Abstract | Crossref Full Text | Google Scholar
10. Hamisch C, Kickingereder P, Fischer M, Simon T, Ruge MI. Update on the diagnostic value and safety of stereotactic biopsy for pediatric brainstem tumors: a systematic review and meta-analysis of 735 cases. J Neurosurg Pediatr. (2017) 20(3):261–8. doi: 10.3171/2017.2.PEDS1665
PubMed Abstract | Crossref Full Text | Google Scholar
11. Lobon-Iglesias MJ, Laurendeau I, Guerrini-Rousseau L, Tauziede-Espariat A, Briand-Suleau A, Varlet P, et al. NF1-like Optic pathway gliomas in children: clinical and molecular characterization of this specific presentation. Neurooncol Adv. (2020) 2(Suppl 1):i98–106. doi: 10.1093/noajnl/vdz054
PubMed Abstract | Crossref Full Text | Google Scholar
12. Pfaff E, El Damaty A, Balasubramanian GP, Blattner-Johnson M, Worst BC, Stark S, et al. Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. Eur J Cancer. (2019) 114:27–35. doi: 10.1016/j.ejca.2019.03.019
PubMed Abstract | Crossref Full Text | Google Scholar
13. Izquierdo E, Carvalho DM, Mackay A, Temelso S, Boult JKR, Pericoli G, et al. DIPG harbors alterations targetable by MEK inhibitors, with acquired resistance mechanisms overcome by combinatorial inhibition. Cancer Discov. (2022) 12(3):712–29. doi: 10.1158/2159-8290.CD-20-0930
PubMed Abstract | Crossref Full Text | Google Scholar
14. Kline C, Jain P, Kilburn L, Bonner ER, Gupta N, Crawford JR, et al. Upfront biology-guided therapy in diffuse intrinsic pontine glioma: therapeutic, molecular, and biomarker outcomes from PNOC003. Clin Cancer Res. (2022) 28(18):3965–78. doi: 10.1158/1078-0432.CCR-22-0803
PubMed Abstract | Crossref Full Text | Google Scholar
15. Karandikar PV, Suh L, Gerstl JVE, Blitz SE, Qu QR, Won SY, et al. Positioning SUMO as an immunological facilitator of oncolytic viruses for high-grade glioma. Front Cell Dev Biol. (2023) 11:1271575. doi: 10.3389/fcell.2023.1271575
留言 (0)