





記住我






Psoriasis is a multisystem, inflammatory skin disease associated with substantial morbidity and mortality (1–3). It is a chronic skin disorder that results in disfigurement, stigmatization, and disability, negatively impacting patient quality of life (4, 5). Further, it is linked to systemic conditions, such as cardiovascular disease, metabolic syndrome, inflammatory bowel disease, psoriatic arthritis, and depression (6, 7). An estimated 2% of the global population has psoriasis, of which 15–20% have severe disease requiring systemic therapy (8).
Systemic treatment options for severe psoriasis include retinoids, traditional immunosuppressants (such as methotrexate or cyclosporine), biologics, and oral small molecules (9). Over the past two decades, novel therapeutic agents, such as biologic therapies targeting TNF-α, IL-12/23, IL-17, and IL-23, have revolutionized psoriasis care such that near-total or total skin clearance has become the gold standard outcome measure used to assess treatment efficacy. However, these agents may be associated with side effects that negatively impact patient treatment adherence and/or persistence. In many instances, direct attribution and assessment of adverse event (AEs) causality can be difficult.
The nocebo effect is a well-established phenomenon defined as the occurrence of undesirable side effects secondary to negative patient expectations as opposed to the pharmacologic activity of an intervention (10–13). For example, Napadow et al. (14) previously demonstrated that patients with atopic dermatitis who anticipated exposure to an allergen reported increased itch with a control saline prick compared to those without similar preconceptions. The nocebo effect has important implications for both research and clinical care by limiting the accurate identification of treatment-emergent AEs in randomized controlled trials (RCTs), thereby increasing treatment-unrelated AEs in intervention arms, placebo arms, or both; resulting in the premature discontinuation of appropriate therapy, leading to increased disease burden and accumulation of disease-specific complications; and negatively influencing the patient-provider therapeutic relationship, reducing patient trust in selected medication options and impacting the provider’s approach to medication counseling.
To date, nocebo effects in psoriasis have not been thoroughly studied. Therefore, we performed a systematic review and meta-analysis of placebo-controlled RCTs of systemic therapies for moderate-to-severe plaque psoriasis with two objectives: to estimate the pooled proportion of patients randomized to placebo who experienced AEs, serious AEs (SAEs), AEs resulting in treatment discontinuation, infections, and injection- or infusion-related AEs; and to characterize the risk differences (RDs) in these outcomes between patients randomized to investigational product versus placebo, stratified by treatment class. Topical therapies were excluded from this review and meta-analysis to provide focus and depth on the exciting and rapidly growing market of systemic psoriasis treatments.
Materials and methodsThe Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines was employed (15).
Search strategyA living systematic review and network meta-analysis by the Cochrane Library has compiled phase II–IV RCTs of systemic therapies in adult patients with moderate-to-severe plaque psoriasis through to October 2021 (9). Eligible studies from this living review were included for analysis. The data was supplemented by searching Embase (Ovid), MEDLINE (Ovid), and the Cochrane CENTRAL Register of Controlled Trials up to January 1st, 2023. The full search strategy is outlined in Supplementary Table 1 and includes terms to capture psoriasis, systemic therapy, and placebo-controlled trials. References of relevant publications were also screened, and only studies published in the English language were included.
Study selectionStudies were included for analysis using the following criteria: placebo-controlled phase II, III, or IV induction or maintenance clinical trials of patients aged ≥18 with moderate-to-severe plaque psoriasis; evaluation of conventional systemic anti-psoriatic agent [defined as methotrexate, cyclosporine, oral retinoid, fumaric acid ester, biologic, and/or oral small molecule (apremilast or deucravacitinib)] versus placebo; and published frequency and nature of AEs (including any AE, SAE, AE requiring treatment discontinuation, infections, and/or injection- or infusion-related AE) in both treatment and placebo groups. Phase I clinical trials were excluded given substantial methodological differences compared to phase II to IV studies.
All citations were independently reviewed by two separate investigators (BM and Y-JP) using the above predefined inclusion criteria. Studies were screened by title and abstract followed by full text review. Disagreements were settled by a third author (PM). All screening was performed using Covidence Systematic Review software (Veritas Health Innovation, Melbourne, Australia).
Outcomes and data extractionThe primary outcome was the pooled proportion of patients experiencing any AE in the placebo arm, and associated RD between systemic therapy and placebo. Secondary outcomes included pooled RDs in SAEs, AEs requiring treatment discontinuation, infections, and infusion- or injection-related AEs between treatment and placebo groups. All AEs were defined and reported by the original study authors. For trials testing multiple interventions, the proportion of patients with each outcome was pooled by treatment class. Multiple doses of systemic therapy were pooled if applicable. Data only over the initial placebo-controlled portion of trials were included.
Trial features that were extracted included: study design and setting (phase, number of centers, duration of follow-up); psoriasis severity criteria; number of patients randomized to systemic therapy and placebo; and incidence and nature of AEs in both intervention and treatment arms. The Cochrane Collaboration Risk of Bias tool version 2.0 was used to assess the methodological quality of included trials (16).
Statistical analysisThe proportion of patients experiencing the primary and secondary outcomes in each of the placebo and active treatment arms were pooled using a random effects model to account for between- and within-study heterogeneity. The Freeman-Tukey double arcsine transformation was used to compute 95% confidence intervals (CI) using the score statistic and exact binomial method. The pooled RD between placebo and intervention arms stratified by medication class (biologic versus non-biologic) were calculated using a restricted maximum likelihood random effects model with 95% CIs. Statistical heterogeneity was quantified using Cochran’s Q and I2 statistics, and interpreted based on Cochrane recommendations (I2 = 30–60% representing moderate, 50–90% substantial, and 75–100% considerable heterogeneity). Univariate meta-regression was used to explore potential causes of heterogeneity using the variables of publication year, trial phase, multinational versus single country study, number of trial centers, and medication class. Publication bias was assessed using funnel plots and Egger’s test. All analyses were performed in Review Manager 5.4 and Stata 17.0 using the metaprop program (StataCorp LLC, College Station, TX).
Results Search results and included studiesThe final analysis included 103 unique RCTs representing 92 comparisons of biologic therapies and 38 comparisons of non-biologic treatments versus placebo (Figure 1 and Supplementary Appendix 1), enrolling a total of 30,249 patients randomized to systemic therapy (25,067 biologic, 82.9%) and 12,940 patients randomized to placebo. Ninety-six trials had initial placebo-controlled periods of 16 weeks or shorter. Four clinical trials comparing acitretin versus placebo were excluded from the analysis due to an inability to locate and confirm the primary outcomes (17–20). Any AE was reported in 96 comparisons (86 biologic and 10 non-biologic), SAEs in 110 comparisons (91 biologic and 19 non-biologic), AE leading to discontinuation of therapy in 107 comparisons (87 biologic and 20 non-biologic), and infectious AE in 101 comparisons (89 biologic and 12 non-biologic). A total of 52 comparisons of biologic agents reported either injection or infusion-related AEs. There was good agreement between reviewers on final studies for inclusion (Cohen’s Kappa 0.58, 93.8% agreement). Most trials were considered at low risk of randomization, missing data, and reporting bias (Supplementary Tables 2, 3).
Figure 1. PRISMA flow diagram.
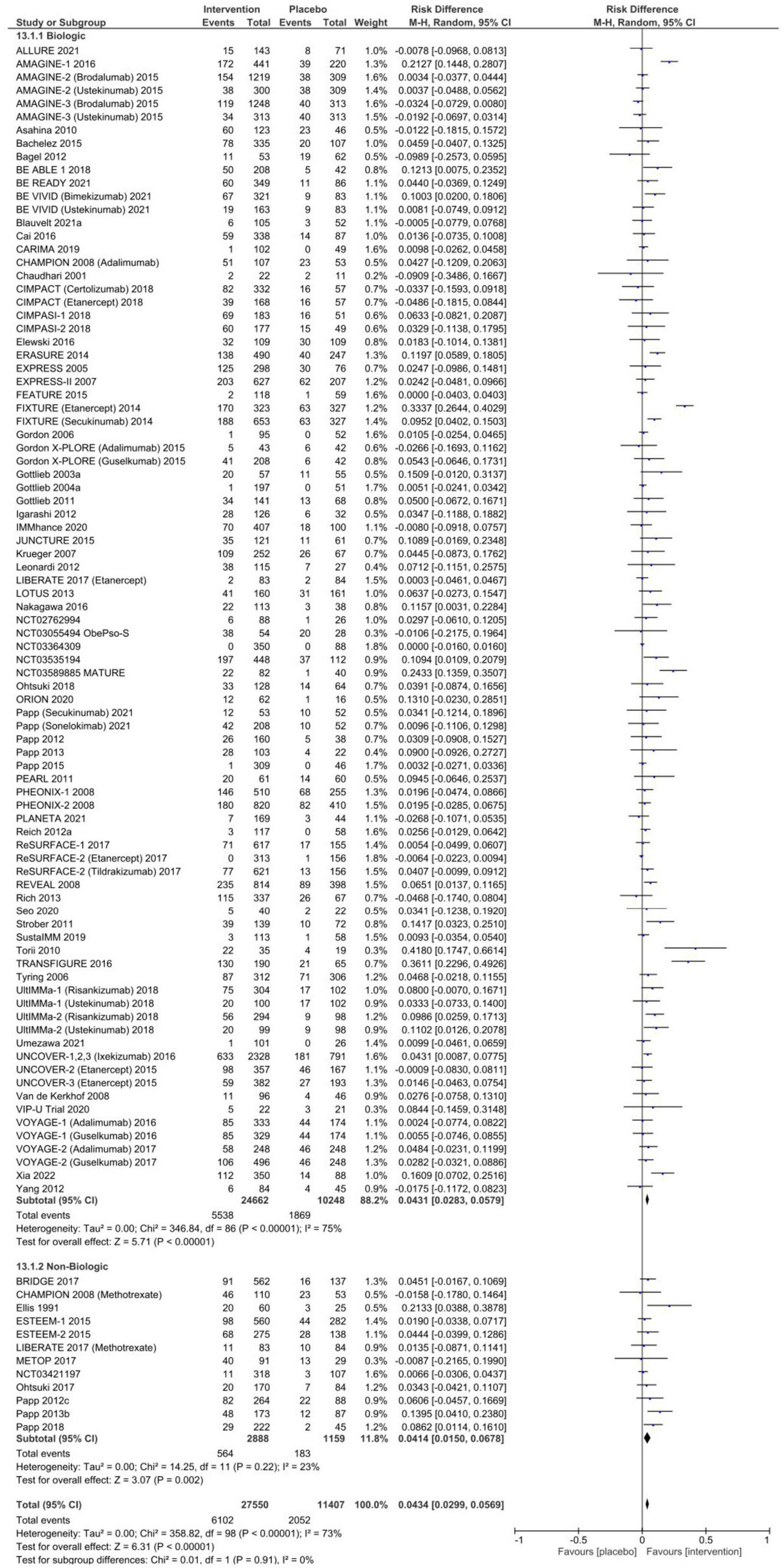
Risk difference in AEsThe pooled RD for any AE between systemic therapy and placebo is summarized in Figure 2 and Table 1. A total of 49.8% [95% CI: 47.1–52.4%] of patients randomized to placebo experienced an AE, compared to 57.1% [95% CI: 54.7–59.5%] in systemic therapy groups, resulting in a RD of 6.7% [95% CI: 4.6–8.9%, p < 0.00001] with considerable overall heterogeneity (I2 = 75%). This RD was observed in subgroup analyses for both biologic [RD 5.3% (95% CI: 3.0–7.5%), p < 0.00001, I2 = 71%] and non-biologic therapies [RD 12.6% (95% CI 7.3–18.0%), p < 0.00001, I2 = 81%] compared to placebo. There was no evidence of publication bias (Supplementary Figure 1, Egger p-value = 0.45).
Figure 2. Pooled risk difference of any adverse event between patients treated with biologic or non-biologic therapy and placebo.
Table 1. Pooled proportion of patients experiencing adverse events and risk difference between systemic therapy and placebo stratified by treatment class.
The pooled proportion of patients receiving placebo who experienced an SAE was 1.4% [95% CI: 1.2–1.7%], and 1.9% [95% CI: 1.5–2.3%] of placebo patients discontinued therapy due to an AE. No statistically significant risk difference between patients who received systemic therapy and placebo for SAEs [RD 0.3% (95% CI 0.0–0.6%), p = 0.06, Figure 3] or AEs necessitating discontinuation of therapy [RD 0.2% (95% CI −0.2–0.6%), p = 0.28, Figure 4] was observed. In subgroup analysis, 6.5% (95% CI: 4.1–9.3%) of patients exposed to non-biologic agents experienced AEs requiring medication discontinuation compared to 3.6% [95% CI: 2.7–4.7%] of placebo patients (RD 2.9% [95% CI: 0.5–5.2%], p = 0.02) with considerable heterogeneity (I2 = 78%). Subclassification by year of publication, trial phase, multinational or single country, number of centers, and drug class in meta-regression resolved heterogeneity in SAEs (I2 = 14.75%), but only partially explained heterogeneity in AE leading to discontinuation (I2 = 49.50%).
Figure 3. Pooled risk difference of serious adverse event between patients treated with biologic or non-biologic therapy and placebo.
Figure 4. Pooled risk difference of adverse events leading to discontinuation of therapy between patients treated with biologic or non-biologic therapy and placebo.
There was a higher risk of infections in patients receiving systemic therapy compared to placebo [RD 4.3% (95% CI 3.0–5.7%), p < 0.00001] that persisted in subgroup analyses for biologic agents [RD 4.3% (95% CI 2.8–5.8%), p < 0.00001, I2 = 75%, Figure 5] and non-biologic agents [RD 4.1% (95% CI: 1.5–6.8%), p = 0.002, I2 = 23%].
Figure 5. Pooled risk difference of infectious adverse events between patients treated with biologic or non-biologic therapy and placebo.
Injection- and infusion-related AEs are summarized in Supplementary Figure 2. There was a significant increase in the risk of injection or infusion-related AEs in the systemic therapy groups compared to placebo [RD 3.4% (95% CI 1.9–4.9%), p < 0.00001], although with significant heterogeneity (I2 = 89%). A total of 1.7% (95% CI: 1.0–2.5%) of placebo-treated patients experienced an injection or infusion-related AE.
DiscussionIn this systematic review and meta-analysis, we examined over 100 RCTs of systemic therapies for moderate-to-severe plaque psoriasis in more than 30,000 patients and identified several key findings. First, nearly 50% of patients receiving inert placebo experienced an AE. This high baseline rate of AEs may be partially explained by nocebo effects and has important implications for RCT design and for evaluating side effects in clinical care. Second, there were statistically significant but numerically low increased rates of all AEs and infections in trial participants receiving systemic therapies for psoriasis compared to placebo. However, we did not observe any significant difference in the proportion of placebo-treated compared to patients receiving systemic therapy who experienced SAEs or required treatment discontinuation, which should inform discussions with patients when starting systemic therapy.
Research in context of existing literatureRandomized controlled trials (RCTs) of biologic and non-biologic agents for psoriasis have consistently identified non-specific medication-exposure related AEs (ex. nausea and headache) prone to nocebo effects (9). These effects have been well characterized in numerous drug classes, including HMG-CoA reductase inhibitors, (21) anti-depressants, (22) anti-epileptics, (23) and biologic therapies for other indications (24). However, there has been limited analysis of its role in systemic agents used for dermatologic indications. The pathogenesis of nocebo responses is complex and multifactorial, encompassing negative patient expectations secondary to perceived sensitivity to therapy, prior treatment experiences, and patient-provider therapeutic relationships (25–28). Furthermore, social conditioning from observed responses in others plays a key role, which is especially relevant given modern mass and social media-facilitated distribution of patient experiences with negative side effects and AEs (29). Therefore, patients with dermatologic conditions may be at high risk for nocebo effects because dermatologic diseases are generally highly visible, distressing, and frequently subject to both personal and peer judgment (i.e., stigmatization) that can predispose to negative interpretations of treatment-related events; and have a chronic, relapsing course such that patients may have negative expectations from failing multiple prior combination treatment regimens consisting of both systemic and topical agents.
Impact of nocebo effectsNocebo effects have important implications for RCT interpretation. First, high rates of AEs in patients randomized to placebo should be used to contextualize the overall safety profile of novel therapies in RCTs. Second, RCTs are essential for determining the efficacy of novel treatments. Still, they are generally under-powered for evaluating safety, especially for rare or serious AEs that require longer follow-up durations and greater patient exposure time, which is difficult to accommodate in most phase II and III induction and 1-year maintenance studies (30, 31). Third, nocebo effects in intervention groups may artificially increase rates of reported AEs and potentially mask the identification of true treatment-emergent AEs. Fourth, nocebo effects may lead to discontinuation of therapy and trial withdrawal, which can confound both evaluations of efficacy and safety if withdrawals occur differentially in treatment and placebo groups (32, 33). This has previously been observed in trials of patients switching from bio-originator to biosimilar TNF antagonists, despite numerous studies demonstrating bio-equivalence and non-inferiority (26). Reassuringly, we identified similar rates of treatment discontinuation between treatment and placebo due to AEs in psoriasis trials. Taken together, these findings highlight the importance of post-marketing drug registries, open-label trial extensions, and integrated safety analyses characterizing long-term safety outcomes.
Recognizing and minimizing nocebo effects in clinical practice may improve patient outcomes and enhance treatment persistence. While it is critical to ensure that all patients starting systemic therapy are informed of the risks and benefits of treatment, several strategies have been proposed to minimize nocebo effects. These include positive framing of side effect profiles, explicit disclosure of possible nocebo effects, standardized approaches to questioning for and measuring patient-reported AEs, and authorized concealment of limited disclosure of potentially rare or irrelevant AEs (28, 34–36). However, our findings need to be cautiously generalized to real-world practice given that clinical trials often select for an overall healthier patient population, whereas more comorbid patients may be using multiple concomitant therapies and be intrinsically at higher risk of AEs; and clinical constraints may alter the informed consent process and presentation of potential side effects compared to a controlled trial setting. The treatment context of an RCT itself may influence patient reporting of AEs because the processes of randomization, informed consent, and blinding have all been linked to nocebo effects (28, 37–39). For example, inclusion of possible gastrointestinal upset in written consent forms for unstable angina therapy was shown to increase the proportion of patients withdrawing from the study due to subjective, minor gastrointestinal symptoms by sixfold (39).
Strengths and limitationsOur study has several important strengths. This systematic review and meta-analysis uniquely assesses the pooled proportion of patients experiencing AEs and associated RD between placebo and both biologic and non-biologic therapies in over 30,000 psoriasis patients. However, our study has some key limitations. First, alternative explanations for the high rate of AEs observed in placebo groups should also consider the accumulation of psoriatic complications from the natural history of progressive, untreated disease; potential misattribution of symptoms from related, comorbid psoriatic conditions such as psoriatic arthritis; and potential effects of concomitant topical or systemic therapies for either psoriasis or an associated condition. Second, there was significant heterogeneity between studies when assessing for any AE that was not fully explained in meta-regression. This may be a consequence of differences in defining and reporting AEs, patient characteristics (such as psoriatic involvement of special sites, concomitant psoriatic complications), and/or intervention differences between sub-therapy classes. For example, oral non-biologic medications, like cyclosporine and acitretin, have different mechanisms of action, resulting in potentially distinct side effect profiles.
In conclusion, we performed a comprehensive systematic review and meta-analysis of systemic therapies for moderate-to-severe plaque psoriasis. Nearly half of all patients randomized to placebo experienced AEs. Our evaluation reveals the necessity of considering nocebo effects to account for these findings. We did not identify any significant overall RD in either serious AE or AE leading to discontinuation of therapy between systemic therapy and placebo. These outcomes inform the interpretation of RCT data and influence clinician-patient communication.
Data availability statementThe original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributionsBM: Writing – review & editing, Writing – original draft, Visualization, Validation, Software, Methodology, Investigation, Formal Analysis, Data curation, Conceptualization. Y-JP: Writing – review & editing, Writing – original draft, Methodology, Investigation, Data curation. KB: Writing – review & editing, Writing – original draft. PRM: Visualization, Validation, Supervision, Software, Resources, Project administration, Methodology, Investigation, Funding acquisition, Formal Analysis, Data curation, Conceptualization, Writing – review & editing, Writing – original draft.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2024.1373520/full#supplementary-material
References1. Abuabara K, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Cause-specific mortality in patients with severe psoriasis: a population-based cohort study in the U.K. Br J Dermatol. (2010) 163:586–92. doi: 10.1111/j.1365-2133.2010.09941.x
PubMed Abstract | Crossref Full Text | Google Scholar
2. Horreau C, Pouplard C, Brenaut E, Barnetche T, Misery L, Cribier B, et al. Cardiovascular morbidity and mortality in psoriasis and psoriatic arthritis: a systematic literature review. J Eur Acad Dermatol Venereol. (2013) 27, (Suppl. 3):12–29.
3. Nijsten T, Wakkee M. Complexity of the association between psoriasis and comorbidities. J Invest Dermatol. (2009) 129:1601–3.
4. Langley RG, Krueger GG, Griffiths C. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. (2005) 64 Suppl 2, (Suppl. 2):ii18–23.
5. Griffiths CE, Armstrong AW, Gudjonsson JE, Barker J. Psoriasis. Lancet. (2021) 397:1301–15.
6. Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. (2017) 76:377–90.
8. Parisi R, Iskandar IY, Kontopantelis E, Augustin M, Griffiths CE, Ashcroft DM, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. (2020) 369:m1590. doi: 10.1136/bmj.m1590
PubMed Abstract | Crossref Full Text | Google Scholar
9. Sbidian E, Chaimani A, Garcia-Doval I, Doney L, Dressler C, Hua C, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochr Datab Syst Rev. (2022) 5:CD011535.
10. Isawa M, Kajiyama M, Tominaga Y, Nakada H, Aomori T, Mochizuki M. Review of clinical studies on the nocebo effect. Pharmazie. (2020) 75:548–53.
11. Bagaric B, Jokic-Begic N, Sangster Jokic C. The nocebo effect: a review of contemporary experimental research. Int J Behav Med. (2022) 29:255–65. doi: 10.1007/s12529-021-10016-y
PubMed Abstract | Crossref Full Text | Google Scholar
12. Sonthalia S, Sahaya K, Arora R, Singal A, Srivastava A, Wadhawan R, et al. Nocebo effect in dermatology. Indian J Dermatol Venereol Leprol. (2015) 81:242–50.
13. Colloca L, Miller FG. The nocebo effect and its relevance for clinical practice. Psychosom Med. (2011) 73:598–603.
14. Napadow V, Li A, Loggia ML, Kim J, Mawla I, Desbordes G, et al. The imagined itch: brain circuitry supporting nocebo-induced itch in atopic dermatitis patients. Allergy. (2015) 70:1485–92. doi: 10.1111/all.12727
PubMed Abstract | Crossref Full Text | Google Scholar
15. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. [The PRISMA 2020 statement: an updated guideline for reporting systematic reviewsDeclaracion PRISMA 2020: una guia actualizada para la publicacion de revisiones sistematicas]. Rev Panam Salud Publica. (2022) 46:e112. doi: 10.26633/RPSP.2022.112
PubMed Abstract | Crossref Full Text | Google Scholar
16. Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. (2019) 366:l4898.
17. Sommerburg CK, Eichelberg D, Goos M, Heese A, Holzle E, Koßmann E, et al. Acitretin in combination with PUVA: a randomized double-blind placebo-controlled study in severe psoriasis. J Eur Acad Dermatol Venereol. (1993) 2:308–17.
18. Goldfarb MT, Ellis CN, Gupta AK, Tincoff T, Hamilton TA, Voorhees JJ. Acitretin improves psoriasis in a dose-dependent fashion. J Am Acad Dermatol. (1988) 18(4 Pt. 1):655–62. doi: 10.1016/s0190-9622(88)70086-9
PubMed Abstract | Crossref Full Text | Google Scholar
19. Saurat JH, Geiger JM, Amblard P, Beani JC, Boulanger A, Claudy A, et al. Randomized double-blind multicenter study comparing acitretin-PUVA, etretinate-PUVA and placebo-PUVA in the treatment of severe psoriasis. Dermatologica. (1988) 177:218–24. doi: 10.1159/000248567
PubMed Abstract | Crossref Full Text | Google Scholar
20. Yilmaz EY, Yerekabakan O. Re-PUVA therapy for psoriasis vulgaris: an effective choice. Turkiye Klinikleri Dermatoloji Dergisi. (2002) 12:204–8.
21. Krishnamurthy A, Bradley C, Ascunce R, Kim SM. SAMSON and the nocebo effect: management of statin intolerance. Curr Cardiol Rep. (2022) 24:1101–8.
22. Rutherford BR, Wall MM, Glass A, Stewart JW. The role of patient expectancy in placebo and nocebo effects in antidepressant trials. J Clin Psychiatry. (2014) 75:1040–6.
23. Zaccara G, Giovannelli F, Giorgi FS, Franco V, Gasparini S. Analysis of nocebo effects of antiepileptic drugs across different conditions. J Neurol. (2016) 263:1274–9. doi: 10.1007/s00415-015-8018-7
PubMed Abstract | Crossref Full Text | Google Scholar
25. Colloca L, Barsky AJ. Placebo and nocebo effects. N Engl J Med. (2020) 382:554–61.
26. Rossettini G, Camerone EM, Carlino E, Benedetti F, Testa M. Context matters: the psychoneurobiological determinants of placebo, nocebo and context-related effects in physiotherapy. Arch Physiother. (2020) 10:11. doi: 10.1186/s40945-020-00082-y
PubMed Abstract | Crossref Full Text | Google Scholar
27. Frisaldi E, Shaibani A, Benedetti F. Understanding the mechanisms of placebo and nocebo effects. Swiss Med Wkly. (2020) 150:w20340.
28. Wartolowska K. The nocebo effect as a source of bias in the assessment of treatment effects. F1000Res. (2019) 8:5.
29. MacKrill K, Gamble GD, Petrie KJ. The effect of television and print news stories on the nocebo responding following a generic medication switch. Clin Psychol Eur. (2020) 2:e2623. doi: 10.32872/cpe.v2i2.2623
PubMed Abstract | Crossref Full Text | Google Scholar
30. Tsang R, Colley L, Lynd LD. Inadequate statistical power to detect clinically significant differences in adverse event rates in randomized controlled trials. J Clin Epidemiol. (2009) 62:609–16.
31. Phillips R, Hazell L, Sauzet O, Cornelius V. Analysis and reporting of adverse events in randomised controlled trials: a review. BMJ Open. (2019) 9:e024537.
32. Pouillon L, Socha M, Demore B, Thilly N, Abitbol V, Danese S, et al. The nocebo effect: a clinical challenge in the era of biosimilars. Expert Rev Clin Immunol. (2018) 14:739–49. doi: 10.1080/1744666X.2018.1512406
PubMed Abstract | Crossref Full Text | Google Scholar
33. Tweehuysen L, van den Bemt BJ, van Ingen IL, de Jong AJ, van der Laan WH, van den Hoogen FH, et al. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthr Rheumatol. (2018) 70:60–8. doi: 10.1002/art.40324
PubMed Abstract | Crossref Full Text | Google Scholar
35. Manai M, van Middendorp H, Veldhuijzen DS, Huizinga TW, Evers AW. How to prevent, minimize, or extinguish nocebo effects in pain: a narrative review on mechanisms, predictors, and interventions. Pain Rep. (2019) 4:e699.
36. Sanderson C, Hardy J, Spruyt O, Currow DC. Placebo and nocebo effects in randomized controlled trials: the implications for research and practice. J Pain Symptom Manage. (2013) 46:722–30. doi: 10.1016/j.jpainsymman.2012.12.005
PubMed Abstract | Crossref Full Text | Google Scholar
37. Silvestri A, Galetta P, Cerquetani E, Marazzi G, Patrizi R, Fini M, et al. Report of erectile dysfunction after therapy with beta-blockers is related to patient knowledge of side effects and is reversed by placebo. Eur Heart J. (2003) 24:1928–32. doi: 10.1016/j.ehj.2003.08.016
PubMed Abstract | Crossref Full Text | Google Scholar
38. Bartley H, Faasse K, Horne R, Petrie KJ. You can’t always get what you want: the influence of choice on nocebo and placebo responding. Ann Behav Med. (2016) 50:445–51. doi: 10.1007/s12160-016-9772-1
PubMed Abstract | Crossref Full Text | Google Scholar
39. Myers MG, Cairns JA, Singer J. The consent form as a possible cause of side effects. Clin Pharmacol Ther. (1987) 42:250–3.
留言 (0)