
記住我







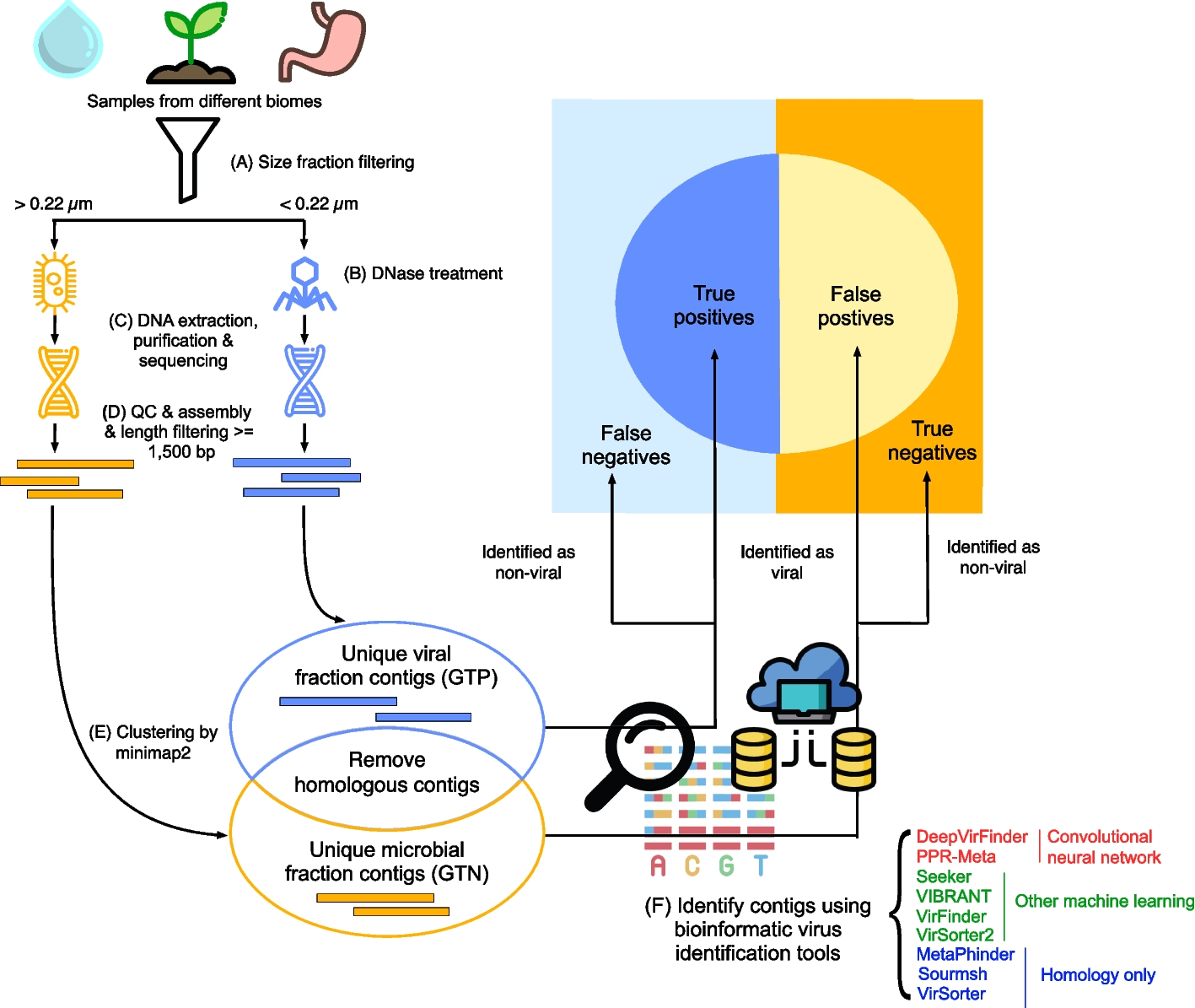


Briefly, the single-cell analysis pipeline with DANCE platform includes data collection, data downloading, data processing (preprocessing and graph construction), and model development on specific downstream tasks (Fig. 1a).
Fig. 1
User perspective of DANCE platform. a Overview of single-cell omics analysis pipeline with DANCE platform. Benchmark datasets by the task are organized and cached on the cloud in advance for users’ usage. Those data cover scRNA-seq data, multimodal single-cell data like Chromium Single Cell Multiome ATAC + Gene Expression and cellular indexing of transcriptomes and epitopes (CITE-seq), and spatially resolved transcriptomic data. After automatic data downloading from the cloud, the DANCE built-in preprocessing and graph construction (required for graph neural networks model development) are then executed. Subsequently, users can build up their own models via customized deep learning model module in DANCE or utilize our reimplemented state-of-the-art deep learning models in DANCE to accomplish downstream tasks. b Currently supported downstream tasks in DANCE
Benchmark dataset collectionWe first collect standard and popular benchmark datasets for each supported downstream task in DANCE. Then, those datasets are organized and cached by dataset name on the cloud.
Data downloadingFor each task, DANCE provides a generic interface to load datasets. Since all benchmark datasets supported by DANCE are cached on the cloud in advance, users do not have to download their interested datasets manually. They just need to specify a dataset name when calling the data loader interface. For example, we can run graph-sc model on 10X PBMC dataset for the clustering task using the following command line:
After data loading, a collection of data processing methods are provided before model training. They are divided into two parts: preprocessing and graph construction.
Preprocessing: We provide rich preprocessing functions such as normalization, dimension reduction, and gene filtering. Take graph-sc model as an example, we filter out the rarely expressed genes and normalize the remaining to obtain the same total count for each cell. Then, only the highly expressed genes (top 3000 by default) are kept for clustering [25].
Graph construction: This is required for GNN-based method. Before model training, we have to convert data to graphs in preparation for graph operations. DANCE provides a variety of ways of graph construction. In graph-sc implementation, we construct a weighted heterogeneous cell-to-gene graph, where the types of nodes can be cell and gene nodes. There are weighted edges between cell nodes and the expressed gene nodes. Let the raw data cell-gene matrix be X, then the weight of gene i to cell j is \(w_=\frac^m X[k,j]}\). There is no edge linked between any pairs of cell or gene nodes.
Model developmentAll types of deep learning models by task have been reimplemented in DANCE with a generic backend framework and unified interface for usage. Users can directly apply them to their interested downstream tasks or build up their own model via our provided customized deep learning module in DANCE.
As shown in Fig. 1b, DANCE presently supports tasks of data spanning through single modality profiling, multimodal profiling, and spatial transcriptomics, which correspond to three stages of single-cell technology development. For a single-modal module, only a single modality like gene expression in the cell can be obtained for analysis. Imputation, cell type annotation, and clustering tasks are supported under this module. For the multimodal module, multiple modalities for the cell can be accessed. For example, CITE-seq can provide both gene expression and protein data for analysis. Modality prediction, modality matching, and joint embedding are currently supported. For the spatial transcriptomics module, the spatial location of the cell in the tissue can be obtained additionally. Spatial domain identification and cell type deconvolution are presently placed under this module. For more details about each task, please refer to the “Methods” section.
Deep learning libraryWe have seen the rapid development of deep learning in single-cell analysis in recent years [32,33,34,35,36,37,38,39] due to its capability of handling huge, high-dimensional, and sparse data. Among them, GNN, as a branch of deep learning, is playing an increasingly important role in the filed of single-cell analysis [25, 26, 40,41,42,43,44] because it is natural to represent cell-gene in a graph, include prior knowledge into graphs, and extract gene-gene patterns hidden from cell-gene relations via propagation. To facilitate the development of deep learning models in this field, we not only provide all kinds of basic deep learning model implementations like commonly used autoencoders (AEs) [45], generative adversarial networks (GANs) [46], and convolutional neural network (CNN) [47, 48] but also support all types of graph operations like graph convolutional network (GCN) [49] and graph attention network (GAT) [50]. What is more, due to the fact that the original single-cell data is not a graph, we also design several interfaces for users to construct various graphs, like cell-cell, cell-gene, and gene-gene graphs after which one of the GNNs is applied.
Benchmark overview: modules, tasks, models and benchmark datasetsAs shown in Fig. 2, DANCE is capable of supporting modules of single modality, multimodality, and spatial transcriptomics. Under each module, we benchmark several tasks with popular models across standard datasets.
Fig. 2
A summary of modules, tasks, models, and datasets supported by the DANCE package
Here, we take the task of clustering in the module of single modality as an example. Various types of methods are implemented including GNN-based methods including graph-sc [25], scTAG [51], and scDSC [35] and AE-based methods including scDeepCluster [34] and scDCC [52]. To ensure a systematic evaluation and fair performance comparison of different models, several standard benchmark datasets such as 10X PBMC 4K [53], Mouse Bladder Cells [54], Worm Neuron Cells [55], and Mouse Embryonic Stem Cells [56] for the task are collected for evaluation. Currently, there are 3 modules, 8 tasks, 32 models, and 21 datasets supported by DANCE. Please refer to the “Methods” section for more details about supported models and datasets.
Comparison with existing packages for single-cell analysisDANCE is not only acting as a deep learning library to facilitate users’ model development but also as a benchmark platform for comprehensive evaluation. Table 1 summarizes the key differences between DANCE and existing single-cell libraries and toolkits. The highlights of DANCE are summarized as follows:
Comprehensive module coverage: Squidpy [57] proposes an efficient and scalable infrastructure only for spatial omics analysis. DeepCell [58] forms a deep learning library for single-cell analysis but only biological images are covered. The library specializes in models for cell segmentation and cell tracking. Even though the popular Scanpy [59] provides a powerful tool for single-cell analysis spanning all modules, it focuses on the field of data preprocessing instead of modeling. Similarly, even though Seurat [10] touches on all three modules, its R language-based interface restricts its applicability for the development of deep learning methods due to limited R interface support within the deep learning community. Instead, DANCE supports all types of data preprocessing and modeling across all modules including single modality, multimodality, and spatial transcriptomics.
Deep learning infrastructure: With the great increase in the number of single cells, classical methods [60, 61] cannot effectively enjoy the benefit from big single-cell data, while deep learning has been proven to be effective. Furthermore, deep learning techniques are also good at handling high dimensional data, which is common for single-cell data. Unfortunately, the backend framework of the well-known Seurat is R, which limits its potential in the deep learning community due to restricted R interface support in the deep learning community. Scanpy only contains classical methodologies for downstream tasks. Recently, scvi-tools [62] presents a Python library for deep probabilistic analysis of single-cell omics data. With 12 models, scvi-tools offers standardized access to 9 tasks. scvi-tools includes some deep learning methods but lacks the recent GNN-based methods. In terms of models, scvi-tools selects baselines with a concentration on statistical models according to their supporting data protocol. As a comparison, DANCE is a comprehensive deep learning library of single-cell analysis. Popular deep learning infrastructures like AEs [45] and GNNs are supported and applicable for all modules.
Standardized benchmarks: To the best of our knowledge, DANCE is the first comprehensive benchmark platform covering all modules in single-cell analysis. A few unique features have been developed to achieve this goal. We first collect task-specific standard benchmark datasets and provide easy access to them by simply changing the parameter setting. Under each task, representative classical and deep learning algorithms are implemented as baselines. Those baselines are further fine-tuned on all collected benchmark datasets to reproduce similar or even better performance compared to original papers. To easily reproduce the results of our finetuned models, end users only need to run one command line where we wrap all super-parameters in advance to obtain reported performance.
Table 1 Comparison between DANCE and other popular single-cell libraries and toolkitsUnified interfaceAll models in DANCE are reimplemented in a unified development environment based on python language using Pytorch [29], DGL [30], and PyG [31] as backbone frameworks. What is more, all models in DANCE have generic interfaces for usage. As shown in Fig. 3, data loading is executed in a generic way via dataloader.load_data(), and model.preprocessing_pipeline() works for all datasets and models to specify model specific preprocessing functions. The interfaces of data.get_train_data() and data.get_test_data() are used to get training and test data respectively. For model training and evaluation, the unified interface for model training is model.fit(). Furthermore, model.score() acts as a generic interface to evaluate how well each model is. The metric of the score function depends on each task. Take scDeepSort [26] for an example, after fitting the model with chosen hyperparameters, we can access the performance of scDeepSort by calling the score function, which will return accuracy to indicate the quality of cell type annotation as a classification task.
Fig. 3
Consistent user experience
Performance showupTo build up a benchmark platform with the capability of systematic evaluations and fair comparisons of available methods, we first collect standard benchmark datasets by task. Then, we reimplement popular existing works for each task in a unified development environment based on the Python programming language with the Pytorch, DGL, and PyG frameworks as the backbone. Finally, we conduct exhaustive experiments of each reimplemented model on collected datasets. The data type supported in DANCE for benchmarking comes from single modality profiling (RNA, protein, and open chromatin) [1,2,3,4,5,6,7,8,9], multimodal profiling [10,11,12,13,14], to spatial transcriptomics [15,16,17,18,19,20,21,22]. Currently, DANCE supports three tasks in the single-modality module, three tasks in the multi-modality module, and two tasks in the spatial transcriptomics module.
Single-modality module―clusteringClustering is a key component of single-cell analysis in the single-modality module. Researchers can distinguish between different cell types or cell type subgroups in the gene expression data using clustering. Adjusted Rand Index (ARI) is employed as an evaluation metric. Three GNN-based methods (graph-sc [25], scTAG [51], scDSC [35]) and two AE-based methods (scDeepCluster [34], scDCC [52]) have been reimplemented under this task. scDSC is deep structural clustering for single-cell RNA-seq data (scRNA-seq) using AEs and GNNs in conjunction. graph-sc and scTAG both convert scRNA-seq data to the cell-to-gene graph as an input for the graph encoder, while scTAG takes topology adaptive graph convolutional network (TAGCN) [63] as the graph encoder. scDeepCluster is a ZINB-based AE method for clustering. Similar model structure to scDeepCluster, scDCC additionally adds pairwise constraints into the loss function. Those five reimplemented models are evaluated on our collected four standard benchmarking datasets, which are 10X PBMC 4K [53], Mouse Bladder Cells [54], Worm Neuron Cells [55], and Mouse Embryonic Stem Cells [56]. There are 4271 cells and 16,653 genes with protocol as 10x Genomics in 10X PBMC 4K dataset, 2746 cells and 20,670 genes with protocol as Microwell-seq in Mouse Bladder Cells dataset, 4186 cells and 13,488 genes with protocol as sci-RNA-seq in Worm Neuron Cells dataset, and 2717 cells and 24,175 genes with protocol as Droplet Barcoding in Mouse Embryonic Stem Cells dataset. Figure 4a shows performance comparison between our implementation and the original implementation of five popular methods on 10X PBMC 4K and Mouse Embryonic Stem Cells datasets. We note that our graph-sc implementation increases slightly from 0.7 to 0.709 and from 0.78 to 0.82 on 10X PBMC 4K and Mouse Embryonic Stem Cells datasets respectively. scDCC performs similarly with the original implementation on the first dataset. On the other hand, we can also observe that our scDeepCluster achieves a similar performance to the original one on the first dataset but gets a worse performance on the second dataset since the variance among random seeds on the second dataset is large. What is more, scTAG in the original paper did not report its performance on both datasets. Instead, to have systematic evaluations, we fill the space of all missing reported performance. For the performance of five methods on more datasets, please refer to Additional file 5.
Fig. 4
Performance comparison between our implementation and original implementation for supported tasks in the single-modality module. DANCE result represents the mean performance across 20 randomly chosen seeds, while the original result refers to the performance directly extracted from the original paper. a Clustering task. b Cell type annotation task. c Imputation task. Note: N/A indicates no performance report from the original paper
Single-modality module―cell type annotationIn the single-modality module, cell type annotation is to annotate the cell type of an individual cell by comparing the query data to annotated reference data (e.g., a single-cell atlas) or employing marker genes indicative of a particular cell type for annotation or modeling as supervised/semi-supervised learning task. Evaluation of model performance is based on prediction accuracy. Five existing works have been reimplemented under this task. scDeepsort [26] is a pre-trained cell type annotation method developed with a weighted GNN framework. Celltypist [64] is a multinomial logistic regression model for classification. SingleCellnet [65] is a random forest-based method, and support vector machine (SVM) [66] is a traditional support vector machine based method to enable the classification of scRNA-seq data. ACTINN [33] is a neural network-based method via multilayer perceptron. Two benchmark datasets have been collected for this task. HCL [67] dataset consists of 562,977 cells, while MCA [68] dataset consists of 201,764 cells. Figure 4b shows performance comparison between our implementation and the original implementation of such five popular methods on the MCA dataset (Mouse Brain 2695 and Mouse Spleen 1759). We can see that most of our implementation models outperform or match the original implementation on both Mouse Brain 2695 and Mouse Spleen 1759 datasets. scDeepsort outperforms the original implementation by a large margin on Mouse Brain 2695 while ACTINN outperforms the original implementation greatly on Mouse Spleen 1759. We also observe that the performance of our scDeepsort and ACTINN is lower than the reported performance from the paper on Mouse Kidney 203 in Additional file 5, which may be explained by the deviation from our implementation or the reported performance from the original paper. For performance comparison on more datasets, please refer to Additional file 5.
Single-modality module―imputationIn the single-modality module, imputation is to correct erroneous zeros by calculating plausible values for gene-cell pairs. For scRNA-seq data, imputation generates false count values for non-expressed genes, but for DNA methylation, imputation provides just the binary one or zero. Mean squared error (MSE) is used as an evaluation metric. Two GNN-based methods and one neural network-based method have been reimplemented under this task. scGNN [40] employs an integrative AE framework that combines gene regulatory signals for scRNA-seq gene expression imputation. GraphSCI [41] employs a graph autoencoder on a cell graph and reconstructs the input using the graph as additional input. DeepImpute [32] constructs multiple neural networks in parallel to infer target genes from an input collection of genes. Four benchmark datasets have been collected for benchmarking under the imputation task. 10X PBMC 5K [69] dataset consists of 5247 cells and 33,570 genes for each cell. Human Embryonic Stem Cells (Human ESC) [70] dataset consists of 758 cells and 17,826 genes for each cell. Mouse Neuron Cells 10k [69] dataset contains 11,843 cells and 31,053 genes for each cell. Mouse ESC [
留言 (0)