
記住我
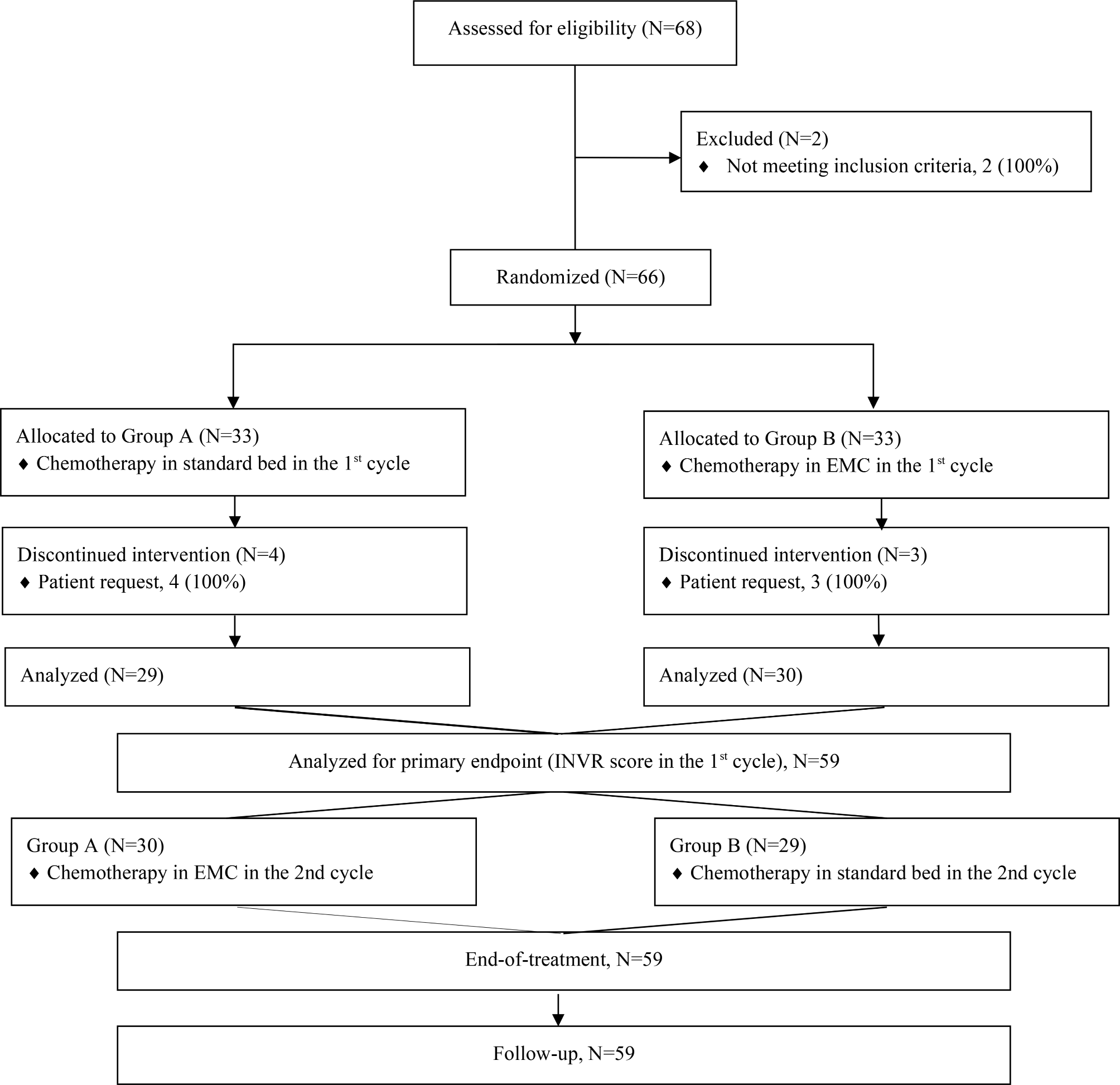
This is a multicenter, randomized, double-blind, placebo-controlled clinical trial with significant advantages in its overall design. After a 7-day screening period, 140 eligible participants will be randomly assigned in a 1:1 ratio to either the trial group (de-walled GLSP + Osimertinib) or the control group (placebo + Osimertinib) through the Interactive Web Response System (IWRS). All participants will undergo two cycles of targeted and experimental drug therapy, followed by a one-month follow-up period. Visits are scheduled on the last day of every 14 days after enrollment. The study is scheduled to be open from February 2023 until February 2026. Recruitment will commence in August 2023 and last for a duration of 18 months (Fig. 1).
Fig. 1
Study process / flow chart
Settings and participatesParticipants will be recruited from thirteen general hospitals located in six different provinces in China, including Beijing Hospital of Traditional Chinese Medicine, Capital Medical University, China-Japan Friendship Hospital, Beijing Friendship Hospital, Capital Medical University, Beijing Chaoyang Hospital, Capital Medical University, Beijing Tongren Hospital, Capital Medical University, Henan Cancer Hospital, the First Affiliated Hospital of Hebei North University, and the First Central Hospital of Baoding City. Additionally, participants will also be recruited from The First Affiliated Hospital of Zhengzhou University, Hubei Provincial Hospital of Traditional Chinese Medicine, Gansu Provincial Cancer Hospital, Beijing Daxing District People’s Hospital and Ningbo Hospital of Traditional Chinese Medicine. Recruitment will be conducted through advertisements or notices posted on the websites and official WeChat accounts of these thirteen hospitals.
The inclusion criteria are as follows: (1) Non-squamous non-small cell lung cancer diagnosed by cytology/histopathology. (2) Clinical staging of inoperable stage IIIA-IV determined through imaging examinations, including chest-enhanced CT, head-enhanced CT/MRI, cervical/supraclavicular lymph node ultrasound/CT, upper abdomen-enhanced CT/B ultrasound, and whole-body bone scan. (3) Driver gene testing with retained tissue specimens after pathological diagnosis or peripheral blood free/tumor DNA (cf/ctDNA) if sufficient tissue specimens cannot be obtained is recommended for epidermal growth factor receptor (EGFR) gene mutations sensitive to 19DEL and 21L858R. (4) Lung lesions must be measurable. (5) ECOG-PS score should not exceed 2 and expected survival should be greater than 3 months. (6) The age range is from 18 to 75 years old. (7) Participants must agree to participate in this study and sign an informed consent form.
The exclusion criteria are as follows:(1) Presence of isolated brain/bone/adrenal metastases to be treated with radiation. (2) Pregnant or lactating women, or with cardiac, pulmonary, hepatic, renal, hematologic, or other systemic severities that are assessed by the investigator to be unsuitable for participation in this study. (3) Known allergy to the study drug. (4) The subject is participating in other clinical trials.
The withdrawal criteria are as follows: (1) Unexpected rapid progression or radiographic confirmation of progression. (2) Complications or coexisting conditions that prevent continued participation in the trial. (3) Serious adverse events occur that make continuation of the trial inappropriate. (4) Erroneous inclusion in the trial was identified. (5) Safety concerns are identified by the investigators. (6) Participants themselves decide to withdraw. Participants have the right to withdraw from the trial during the course of the trial, meanwhile investigators should attempt to identify and document reasons for withdrawal, such as a perceived lack of treatment effect. Some adverse reactions could not be tolerated, unexplained loss to follow-up, etc.
InterventionThe programmatic interventions involve the utilization of Osimertinib, and de-walled GLSP.
Third-generation EGFR-TKI OsimertinibThe initial treatment regimen for EGFR-mutated NSCLC involves the administration of third-generation EGFR-TKI Osimertinib, as recommended by the NCCN Clinical Practice Guidelines for NSCLC 2022 V6. A targeted therapy duration of 56 days will be implemented during the study period.
De-walled Ganoderma Lucidum spore powderAll participants will commence administration on the first day of enrollment and continue for a duration of 2 months. The drugs should be completely dissolved by stirring at a temperature of 100 °C hot water, diluted with cold water or cooled prior to administration two grams twice per day during the initial week, and four grams twice per day from weeks 2 to 8. In parallel, the control group will receive placebo pellets composed of 95% dextrin and 5% de-walled GLSP following an identical protocol. Both TCM granules and placebos are uniformly manufactured by Zhejiang Shouxiangu Pharmaceutical Co., LTD (located at No.10 Shangcheng Road, Hushan Street, Wuyi County, Zhejiang Province) to ensure similarity in terms of appearance, smell, texture, and taste. Participants are required to return the kit along with its label to the CRC as evidence of timely medication usage for adherence evaluation.
Randomization and allocation concealmentThe IWRS of the ProResearchCom Intelligent Monitoring System for Clinical Research will be utilized. Upon enrollment of eligible participants, it is obligatory for the Clinical Research Coordinator (CRC) to input participant details into the IWRS. Once verified by either the Principal Investigators (PI) or Sub-Investigator (Sub-I), participants will be allocated to either the trial or control group in a 1:1 ratio. The IWRS will automatically generate unique identification numbers and drug codes for each participant, subsequently providing feedback on grouping results to the PI or sub-I, who will then notify the drug administrator regarding preparation of drugs corresponding to their respective assigned drug codes.
The current study utilizes a double-blind methodology, whereby the de-walled GLSP drinkable tablets and placebo are enclosed in visually indistinguishable cartons utilizing identical pouches. Each carton is allocated a drug number that conceals details regarding the participant’s group and type. Participants will remain unaware of their treatment allocation until the completion of the trial, while statistical analysts have access to complete group information but do not actively partake in the trial.
Sample size calculationThe modeling calculations were conducted using the PASS 15.0 software, employing a Two-Sample t-test Assuming Equal Variance (bilateral test) with the following parameters: alpha = 0.05, power = 0.90, and N1:N2 = 1:1 ratio. The score of fatigue according to QLQ-C30 is the primary outcome measure of our study. In the FLAURA patient-reported outcome using the same scale, a mean fatigue score of 32.2 ± 24.9 was observed in patients with advanced EGFR-mutated NSCLC treated with Osimertinib [14]. Referring to previous literature, it is considered that the minimum clinically important difference (MID) in fatigue subscale is 11.1 and there exists a range of change between 11.1 and 22.2 points within this threshold value range for clinical significance assessment purposes [15]. The use of de-walled GLSP is assumed to result in at least a 15-point increase in fatigue score, indicating potential clinical significance for this drug intervention approach. By calculating N1 = N2 = 59 cases, we determined that the minimum required sample size would be 118 cases. Considering a dropout rate of 15%, adjusting N1 = N2 = 70 cases yields a minimum sample size requirement of 140 cases.
Assessments and time-pointsAssessments will be conducted biweekly from the beginning of the study until completion (Table 1).
Table 1 Trial visit schedulePrimary outcomeFatigue symptoms as adverse reactions to EGFR-TKIs is the primary outcome of this study, which will be evaluated by the EORTC QLQ-C30 Chinese version. The scale will be administered biweekly throughout the treatment period. The outcome endpoint will be determined based on the QLQ-C30 fatigue subscale score on day 56.
Secondary outcomesThe secondary outcomes are as follows:
The efficacy evaluation of the combination of de-walled GLSP and EGFR-TKIs will be evaluated based on objective response rate (ORR), disease control rate (DCR), and progression-free survival (PFS) according to RECIST 1.1 criteria. Imaging examinations and efficacy assessments will be conducted on day 0, day 42, and day 84 of the treatment period, calculating ORR/DCR, while PFS will be determined by follow-up at each treatment cycle during the follow-up period.
The QoL assessment will be conducted using the QLQ-C30, PFS-CV (Chinese version of the Piper Fatigue Scale), and EQ-5D (European Five Dimensional Health Scale). Participants will independently complete these three questionnaires with assistance from the investigator to evaluate patients’ QoL from a multidimensional perspective. The total score for QLQ-C30 is calculated by summing scores across domains and dividing by the number of items included, which is further linearly transformed using polarization. PFS-CV measures typical fatigue symptoms and is calculated as the mean sum of all topic scores, with subscale scores obtained by averaging four different dimensions separately. EQ-5D quantifies patient health status through five dimensions assessed via a utility index, while a visual scale allows direct scoring by patients on a scale of 0 to 100. QoL will be collected at baseline and each visit.
Exploratory outcomesThis study proposes exploratory indicators and recommends collecting blood for cytokine xii detection, such as IL-1β, IL-2, and TNF-α, and fecal samples for gut microbiota analysis to explore the subsequent mechanisms after obtaining informed consent from participants. All samples will be collected and transported to the central laboratory of Beijing Traditional Chinese Medicine Hospital for unified detection (Figs. 2 and 3).
Fig. 2 Fig. 3
Fig. 3 Safety assessment
Safety assessmentParticipants will undergo regular monitoring of blood, urine, feces, liver and kidney function, as well as electrocardiograms on Day 0 and Day 28 of the treatment period. Adverse events will be classified according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI-CTCAE), version 4.03. Any adverse events, serious adverse events, or adverse drug reactions occurring during the study will be documented with details including severity, relevance to this study, duration, relationship to the trial drug, management measures taken and outcome regression. Follow-up visits will continue until the patient’s condition stabilizes or improves.
Register, ethical issues, and oversightBefore enrolling participants, we registered the study protocol with the China Clinical Trial Registry (ChiCTR2300072786). The study obtained ethical approval from the Ethics Committee of Beijing Hospital of Traditional Chinese Medicine, Capital Medical University (2023BL02-060-01) and other participating institutions. It is currently being conducted in accordance with the principles outlined in the Declaration of Helsinki regarding risks and benefits, privacy and confidentiality, informed consent, and placebo usage. Supervision will be carried out by Beijing Excellence Future International Pharmaceutical Technology Development Co., Ltd (CRO), with clinical research associates (CRAs) independently monitoring safety, quality, and progress on a quarterly basis.
Data managementThe responsibility for data management lies with Beijing Excellence Future International Pharmaceutical Technology Development Co., Ltd. The Data Manager (DM) of the company has implemented Electronic Data Capture (EDC) based on the Case Report Form (CRF) in ProResearch’s Clinical Research Intelligent Monitoring System, following the previously drafted Data Management and Validation Plan. Clinical Research Coordinators (CRCs) can input data into EDC using their accounts. Any errors or queries identified by the system will be forwarded to CRCs for resolution. Once all data entry issues have been resolved, PI, Sub-Is, and CRCs will lock the database within four weeks. Finally, Beijing Excellence Future International Pharmaceutical Technology Development Co., Ltd exports and transfers the locked database to statisticians for analysis purposes. Only the DM has access to the final trial dataset.
Statistical analysisThe data will be subjected to two-sided statistical tests using SAS software (version 9.4). Statistical significance was defined as a P value of 0.05 or less. Following data collection, the Full Analysis set (FAS), per-protocol set (PPS), and safety set (SS) will be established. In order to be included in the per-protocol analysis, participants must provide data on the QLQ-LC30 at baseline and at least two additional time points. If a participant meets the withdrawal criteria, their data will not be included in the analysis. Continuous variables will be presented as mean ± SD or as median and IQR. Comparison between groups will be conducted using t-test or Wilcoxon rank sum test for continuous variables, while categorical variables will be expressed as frequency or proportion and compared using χ2 or Fisher’s exact test between groups. The survival analysis or Logistic model or Cox regression model will then be constructed for multivariate analysis based on these comparisons. Missing data will undergo multiple imputations to handle them appropriately, allowing for sensitivity analyses by comparing results from complete cases with those from imputed data.
留言 (0)