
記住我

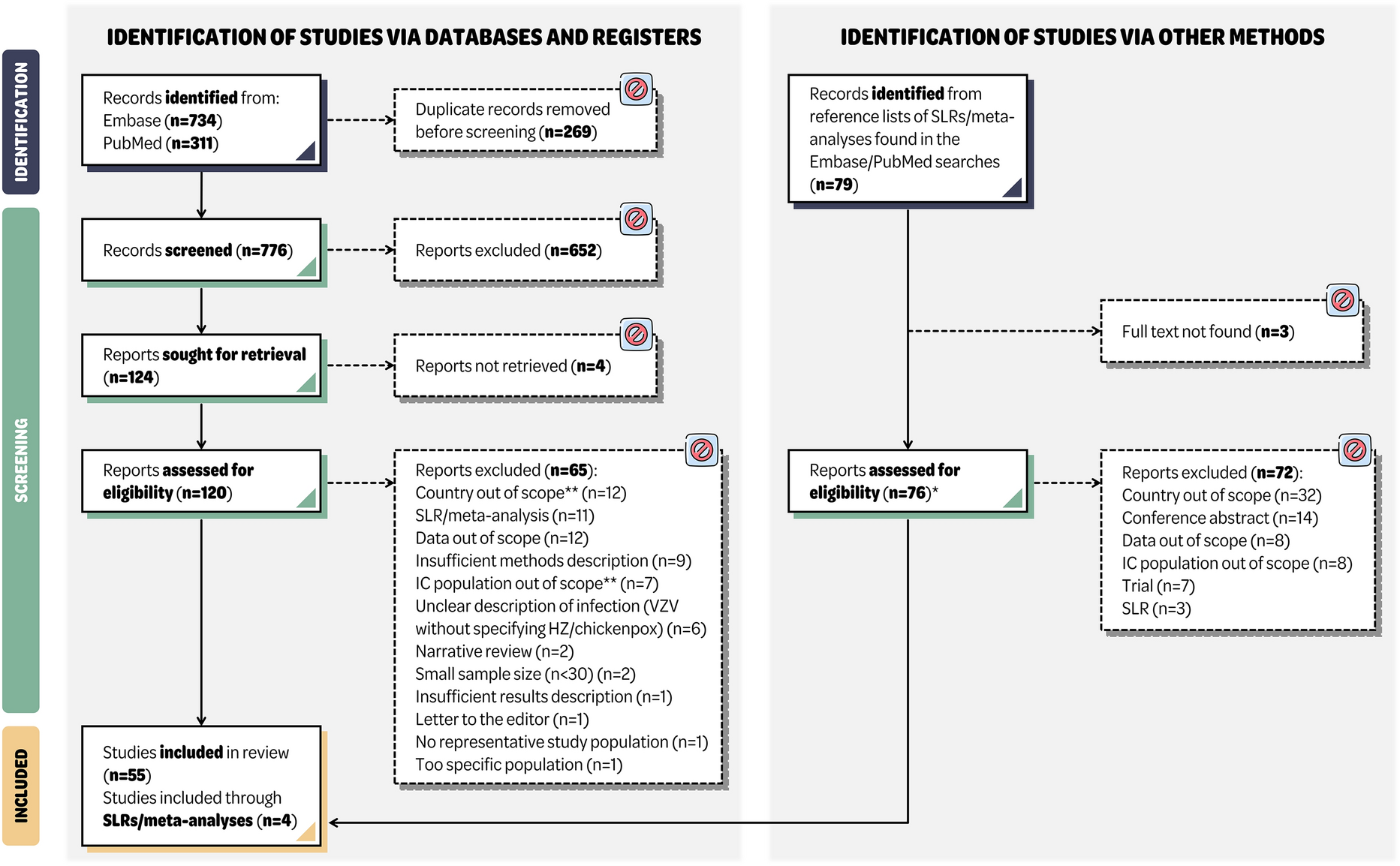







This is a multicenter clinical trial that uses a double-blind, randomized, controlled, parallel-group design with a 1:1 allocation ratio (Fig. 1). The study protocol was formulated in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 guidelines [11] (Table 1). The objective of the trial is to recruit 1240 participants and randomly assign them, using a computer-generated randomization sequence, to either the control group receiving a 96-h infusion set replacement (N = 620) or the treatment group undergoing a 24-h infusion set substitution (N = 620).
Fig. 1
Flow diagram of this trial
Table 1 Standard protocol items: recommendations for interventional trials (SPIRIT) checklistPatient Population and Recruitment StrategyThis study is being conducted in the ICUs of four top-tier tertiary teaching hospitals located in Wuhan, from May 2022 to December 2025: (1) Zhongnan Hospital of Wuhan University (Wuhan, China); (2) Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China); (3) Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China); (4) Renmin Hospital of Wuhan University (Wuhan, China). Among these hospitals, the numbers of ICU beds are 58, 74, 30, and 48 respectively.
Inclusion CriteriaThe study inclusion criteria are as follows:
1.18 years of age or older
2.Capable of providing informed consent
3.Expected to have a length of stay (LOS) in the ICU of more than 96 h
4.In need of treatment with a central venous access device
5.Expected to have the central venous access device in place for at least 96 h with an attached infusion set
Exclusion CriteriaThe study exclusion criteria are as follows:
1.Patients who have a bloodstream infection within 48 h prior to enrollment
2.Patients who have their vascular access device actually removed within 96 h after enrollment
3.Patients who have participated in any other clinical studies within the past 2 months
Sample SizePrevious clinical trial data reported a 3.6% CLABSI incidence rate in the control group [10]. It is hypothesized that a 2.6% absolute difference between the placebo and intervention group will result in a 1% CLABSI incidence in the intervention group. To achieve 80% power and detect a statistically significant difference between the groups with an α value of 0.05, the χ2 test is utilized, and a total of 1160 patients will be needed. Accounting for a 10% exclusion rate due to missing data, our study ultimately will include 1240 patients (620 per group).
Randomization and BlindingOur study plans to randomize patients into two groups, one receiving an infusion set replacement interval of 24 h, and the other receiving an interval of 96 h. The randomization process will be conducted on a per-participant basis using a centralized, computerized service purpose-built by Beijing Medi-Tech Co., Ltd.
To minimize potential bias, strict measures are implemented to limit access to the random sequence solely to the clinical research nurse. The physicians and staff at the study sites are intentionally kept unaware of the hypothesis, group allocation, and study endpoints. Furthermore, the outcome assessors will be blinded to the group assignments as part of the study design, thus ensuring an unbiased evaluation.
ProceduresThe study involves a control group in which infusion devices are replaced every 96 h and an experimental group in which they are replaced every 24 h. Once an eligible patient is identified, a computer platform engineer will randomly assign the patient to either the experimental or control group at a 1:1 ratio. The first day of the study will be designated as the day the vascular access device is inserted, whether for a newly inserted central venous catheter or scheduled surgery with strict aseptic technique in place.
The central venous catheter insertion will be performed in compliance with the Centers for Disease Control (CDC) and EPIC III guidelines, and two trained nursing staff members will complete the preparation and replacement procedures for new infusion devices, with the operating nurse performing the procedure and the research nurse reviewing it, while maintaining hand hygiene. All staff underwent systematic training to minimize the heterogeneity of operations and their impact on the results. Each research unit developed a standardized operating procedure based on the study protocol, and the researcher conducted regular quality control on the operations to minimize heterogeneity. The type and size of vascular access devices will be determined on the basis of the patients’ treatment needs. The Supplementary Material contains the checklist used in this trial.
Patients who have had a central venous access device inserted upon ICU admission must undergo replacement and culturing of the device. Only patients with no detected infection will be eligible to participate in the study, while those with an infection will be excluded. The study will conduct blood, catheter tip, and exudate cultures only when clinical suspicion of infection arises, such as new fever, chills, or hypotension. Cultures will not be routinely performed on asymptomatic patients. The removal of vascular access devices will be based on clinical indications rather than routine removal. Basic characteristics of patients and devices are recorded, and all venous treatments and infusion device replacements are documented by bedside nurses and reviewed by research nurses. Catheter tip sampling for culture involves removing the catheter from the skin-piercing site of the central venous catheter after disinfection with iodine and alcohol, and using sterile scissors to cut it 5 cm away from the catheter tip for bacterial culture and sensitivity testing.
Outcome MeasuresThe primary outcome is the rate of CLABSI within 28 days after randomization. CLABSI is defined as (i) the catheter being in place for at least 48 h before the onset of sepsis, and/or (ii) the presence of microbiologic growth (bacteria and/or fungi) of at least 15 colony forming units (CFU) on the CVC tip that is identical to a positive blood culture sample, and/or (iii) a difference of more than 2 h in time to positivity between a central and a peripheral drawn blood culture. The secondary outcomes of this study include the following:
1.Catheter-related bloodstream infection (CRBSI) rate in the ICU up to day 28, which is defined as a bacteremia or fungemia with clinical manifestations of infection and no other identifiable source, and at least one positive blood culture from a peripheral vein, plus matching organism(s) found on the catheter tip (> 15 CFUs on semiquantitative culture); or, two blood cultures (one from the catheter and one from a peripheral vein) with matching organism(s) that meet the criteria for differential time to positivity (growth of catheter-drawn blood at least 2 h before growth from a peripheral vein blood culture)
2.All-cause bloodstream infection rate in the ICU up to day 28
3.Colonization of the vascular access device in the ICU up to day 4, using the semiquantitative method “Maki roll” and a cutoff of ≥ 15 CFU/catheter
4.Colonization of the infusion set in the ICU up to day 4, using the quantitative broth dilution method and a cutoff of ≥ 1000 CFU/ml
5.ICU all-cause mortality, which is the number of patients who die in the ICU after enrollment in the study
6.In-hospital all-cause mortality, which is the number of patients who die in the hospital after enrollment in the study
7.28-day all-cause mortality, which is the number of patients who die within 28 days after enrollment in the study
8.ICU length of stay
9.Hospital length of stay
10.Cumulative time that the catheter is in situ
11.Number of infusion sets used per patient
12.Costs of consumables for all infusion set replacement procedures per patient
13.Cumulative staff time required for all infusion set replacement procedures per patient
Data CollectionBaseline data will be collected at the time of enrollment, including demographics, comorbidities, reason for ICU admission, length of hospital stay, use of antibiotics, and laboratory values. In addition, data on the incidence of CLABSI, CRBSI, and other relevant outcomes will be collected prospectively during the study period. Patient clinical outcomes will be assessed at 28 days post-enrollment, or until hospital discharge, whichever occurs first.
Safety and MonitoringThis study will administer a variety of health assessments, including an abdominal ultrasound, blood biochemical test, and ECG examination, to provide a comprehensive evaluation of each participant’s health status. Adverse events in this study refer to severe diseases that occur unexpectedly and are clinically significant with respect to replacing the infusion device. These events may be related to the research process, and expected adverse events in the study include all-cause bloodstream infections and death. In case a subject dies or develops a fever with blood test results indicating an infection, the clinical team leader, the responsible person for the project in the unit, the ethics committee, and the safety monitoring committee (composed of epidemiologists responsible for reviewing and evaluating the clinical effectiveness and safety data collected in the study and assessing the accumulated serious adverse events) should be notified within 24 h, and a detailed record should be created. Two trained ICU doctors will determine whether it is an all-cause bloodstream infection (excluding CLABSI). If there is a discrepancy, a third ICU doctor will make the decision. Such subjects must be appropriately handled and followed up until the condition is resolved or stabilizes.
The progress and safety of this trial will be monitored by an independent data and safety monitoring board (DSMB) comprising two academic intensivists with significant experience in conducting clinical trials for critical illnesses. The DSMB has the authority to interrupt the trial to investigate or provide recommendations on potential safety concerns to strengthen the study’s design and implementation.
Quality Control and ManagementPrior to initiating the study, all researchers will undergo a rigorous training program to ensure they possess a comprehensive understanding of the study protocol and standard procedures. Unique numeric identifiers will be assigned to case report forms (CRFs) to precisely record relevant patient data, including adverse events and safety evaluations. Outcome assessors will perform double data entry, and access to the CRFs will be restricted to them. The Evidence-based Medicine Center at Zhongnan Hospital of Wuhan University will diligently oversee and monitor the study’s progress on a quarterly basis.
Statistical AnalysisThe data collected during this study will undergo an intention-to-treat analysis. Demographic characteristics and baseline data will be summarized using descriptive statistics. Binary data will be presented as counts and percentages, and differences between groups will be analyzed using risk differences and risk ratios, along with their corresponding 95% confidence intervals (CIs). The calculation of these intervals will follow the method specified by Miettinen and Nurminen [12]. Continuous data were reported as means with standard deviations or medians with first and third quartiles, depending on their distribution pattern. To detect any differences between groups concerning continuous outcomes, mean differences with 95% CIs were estimated through generalized linear models, which incorporated robust errors for accurate results. The data will undergo time-to-event analysis including all randomized patients. Statistical analysis of the clinical data will be conducted using both the SPSS software program version 24 (SPSS, IBM Corporation) and R statistical software version 3.6.1 (R Foundation), with a significance level set at 0.05, and all hypothesis tests will be two-tailed. Overall, the study adopted a rigorous analytical approach to generate reliable findings.
Ethics and DisseminationThis study has been approved by the institutional review boards and ethics committees of Zhongnan Hospital of Wuhan University (Clinical Ethical Approval No. 2022062). Participants must give informed consent to participate in the study before taking part. This protocol has been registered at the ClinicalTrials.gov, with the ID number of NCT05359601 on 4 May 2022. This study will comply with the principles outlined in the Declaration of Helsinki in 1964 and its subsequent amendments [13].
留言 (0)