Trial Overview
TACKLE was a phase 3, double-blind, placebo-controlled, multicenter study, full details of which have previously been reported [14]. Briefly, eligible participants were adults aged ≥ 18 years who had not received a COVID-19 vaccine and who had laboratory-confirmed SARS-CoV-2 infection determined by reverse transcription-polymerase chain reaction (RT-PCR) or antigen test from any respiratory tract specimen collected within 3 days of enrollment and had a World Health Organization (WHO) Clinical Progression Scale score > 1 and < 4. Participants were randomized 1:1 to receive 600 mg AZD7442 (consecutive intramuscular injections of 300 mg tixagevimab and 300 mg cilgavimab) or placebo within 7 days of symptom onset. The final analysis was performed when those participants who had not withdrawn from the study had completed follow-up through day 457 (approximately 15 months).
Ethical Approval
The study adhered to Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable laws and regulations. The protocol was reviewed and approved by an institutional review board or ethics committee at the respective study sites (Table S1). All participants provided informed, written consent.
Randomization and Masking
Participants were randomly assigned in a 1:1 ratio to either AZD7442 or placebo via a central interactive response system, with stratification by time from symptom onset (≤ 5 or > 5 days) and risk of progression to severe COVID-19 (low or high risk; with high risk including age ≥ 65 years, immunocompromised, and comorbidities such as cancer and chronic diseases). Participants, investigators, and sponsor staff who were involved in treatment, clinical evaluation, and monitoring of the participants were masked to the randomly assigned study treatments.
Endpoints
The primary efficacy endpoint was a composite of severe COVID-19 or death from any cause through day 29, with severe COVID-19 defined as a minimum of pneumonia or hypoxemia together with a WHO Clinical Progression Scale score of ≥ 5. The primary analysis was conducted 30 days after 43 primary endpoint events had occurred. As the primary analysis was conducted whilst the study was ongoing, data continued to accumulate as participants completed protocol visits. Evaluation of the primary efficacy endpoint was repeated at the final analysis (using methods as previously described [14]) to include participants whose day 29 visit came after the primary data cutoff. Also repeated at the final analysis (using methods as previously described [14]) were evaluations of the first supportive estimand of the primary efficacy endpoint, which included participants dosed within 5 days of symptom onset, the key secondary efficacy endpoint, which was a composite of death from any cause or hospitalization for COVID-19 complications or sequelae through day 169, and the levels of SARS-CoV-2 RNA detected in nasal swabs up to day 29.
Safety was evaluated on the basis of incidences of adverse events (AEs), serious AEs (SAEs), and AEs of special interest (AESIs). AESIs included anaphylaxis and any other serious hypersensitivity reactions, injection site reactions, and cardiovascular and thrombotic events.
Single-dose pharmacokinetics of AZD7442 were assessed over the course of the study using methods as previously described [14]. Incidence and serum titers of treatment-emergent antidrug antibodies (TE-ADA) in participants receiving AZD4772, and the impact of TE-ADAs on the pharmacokinetics of AZD7442, were also evaluated (methods as previously described [14]). ADA positivity was defined by ADA titers ≥ 160 for tixagevimab or ≥ 80 for cilgavimab. ADAs were defined as treatment-emergent if they occurred post-dose in participants who were ADA negative at baseline, or if they occurred in participants who were ADA positive at baseline but experienced an increase in ADA titers of ≥ 4-fold over the course of the study.
Protocol Amendments
Amendments to the original study protocol included the redefinition of an AESI to include cardiovascular and thrombotic events, as noted above. A cardiovascular event adjudication committee was commissioned to independently validate the diagnosis of all cardiovascular events, including cardiac ischemic, cardiac failure, cerebrovascular, and thromboembolic AESIs, as well as all fatal AEs (to determine if they were cardiovascular deaths). The protocol amendments also clarified that reinfection/recurrence of COVID-19 or new, confirmed asymptomatic SARS-CoV-2 infections were to be captured as AEs or SAEs and not considered to be the original disease, and that prolonged COVID-19 symptoms lasting approximately 3 months were to be captured as an AE of post-acute COVID-19 symptoms (long COVID-19).
Statistical Analyses
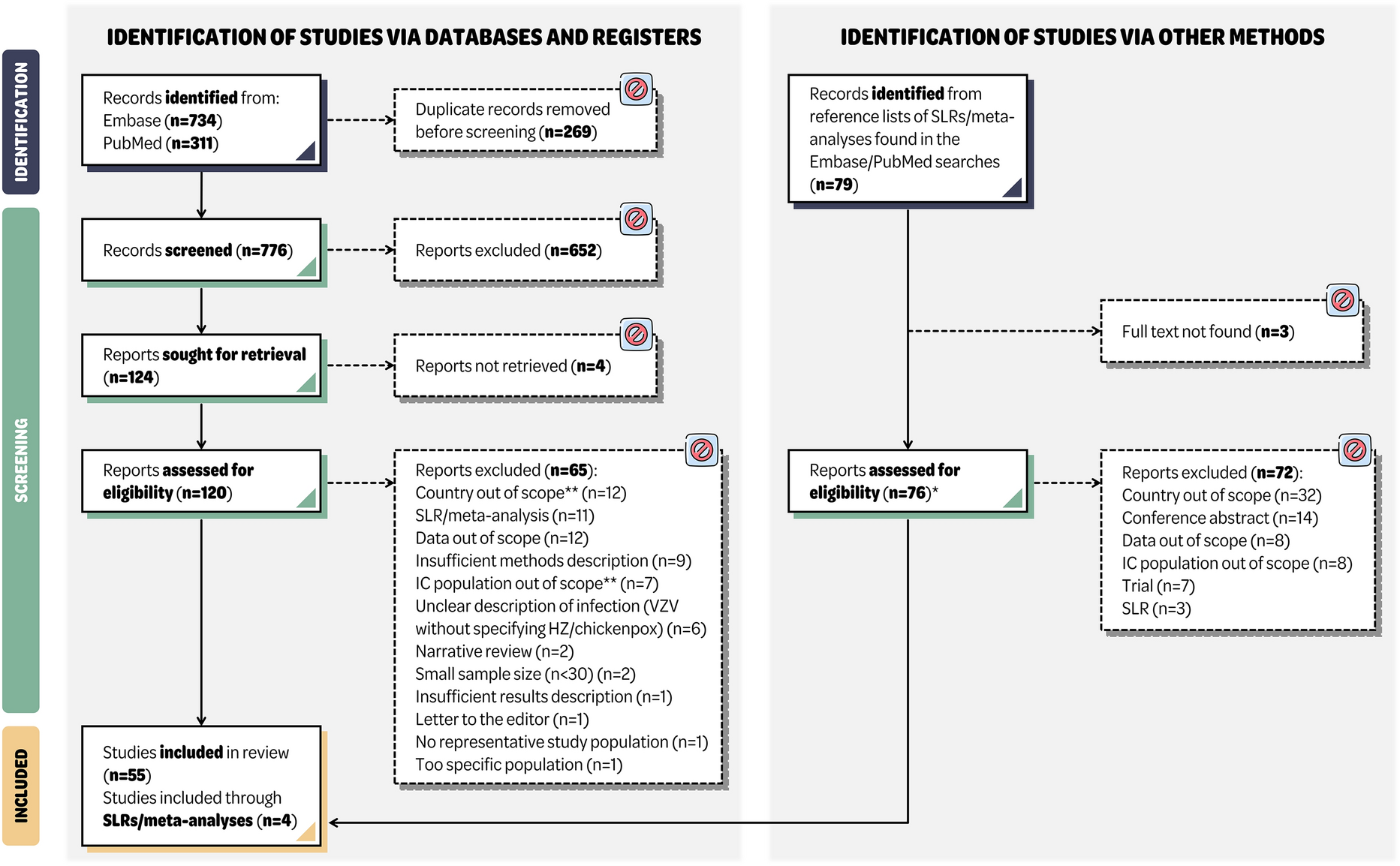
The primary efficacy endpoint was evaluated using the modified full analysis set (mFAS), comprising all participants administered study drug within 7 days of symptom onset and not hospitalized at baseline (day 1) for isolation purposes (hospitalization was allowed for isolation purposes in Japan and Russia as a result of local requirements). The first supportive estimand of the primary efficacy endpoint was evaluated in an early intervention subgroup of participants from the mFAS who were dosed within 5 days of symptom onset. The key secondary endpoint and the change in SARS-CoV-2 RNA from baseline were evaluated in the mFAS. Safety was evaluated using the safety analysis set, comprising all participants who received study drug regardless of baseline hospitalization status. The single-dose pharmacokinetics of AZD7442 were evaluated using the pharmacokinetics analysis set, comprising all participants who received AZD7442 and had at least one quantifiable serum pharmacokinetic observation post-dose. TE-ADAs and their impact on the pharmacokinetics of AZD7442 were evaluated using the AZD7442 ADA set, comprising all participants in the safety analysis set who received AZD7442 and were assessed for ADAs at baseline and at least one post-baseline visit.
The relative risk reduction (RRR) in the primary efficacy endpoint between the AZD7442 and placebo groups was assessed using a Cochran–Mantel–Haenszel test, stratified by the randomization stratification factors. The key secondary efficacy endpoint was assessed by the same methods. Kaplan–Meier curves were used to summarize the time to primary endpoint events. A Cox proportional hazards model used to obtain a hazard ratio (HR) and 95% confidence intervals (CIs), with the stratification factors included as covariates, and a stratified log-rank test was used to assess differences between groups.
The changes from baseline in SARS-CoV-2 RNA levels were compared between groups at day 3 and day 6 using a mixed model for repeated measures. These results were confirmed by calculating the time-weighted average change in log10 SARS-CoV-2 RNA from baseline to day 6 and day 29 from an analysis of covariance model, including terms for log10 baseline value, treatment, randomization stratification factors, and log10 baseline by treatment interaction.









留言 (0)