
記住我










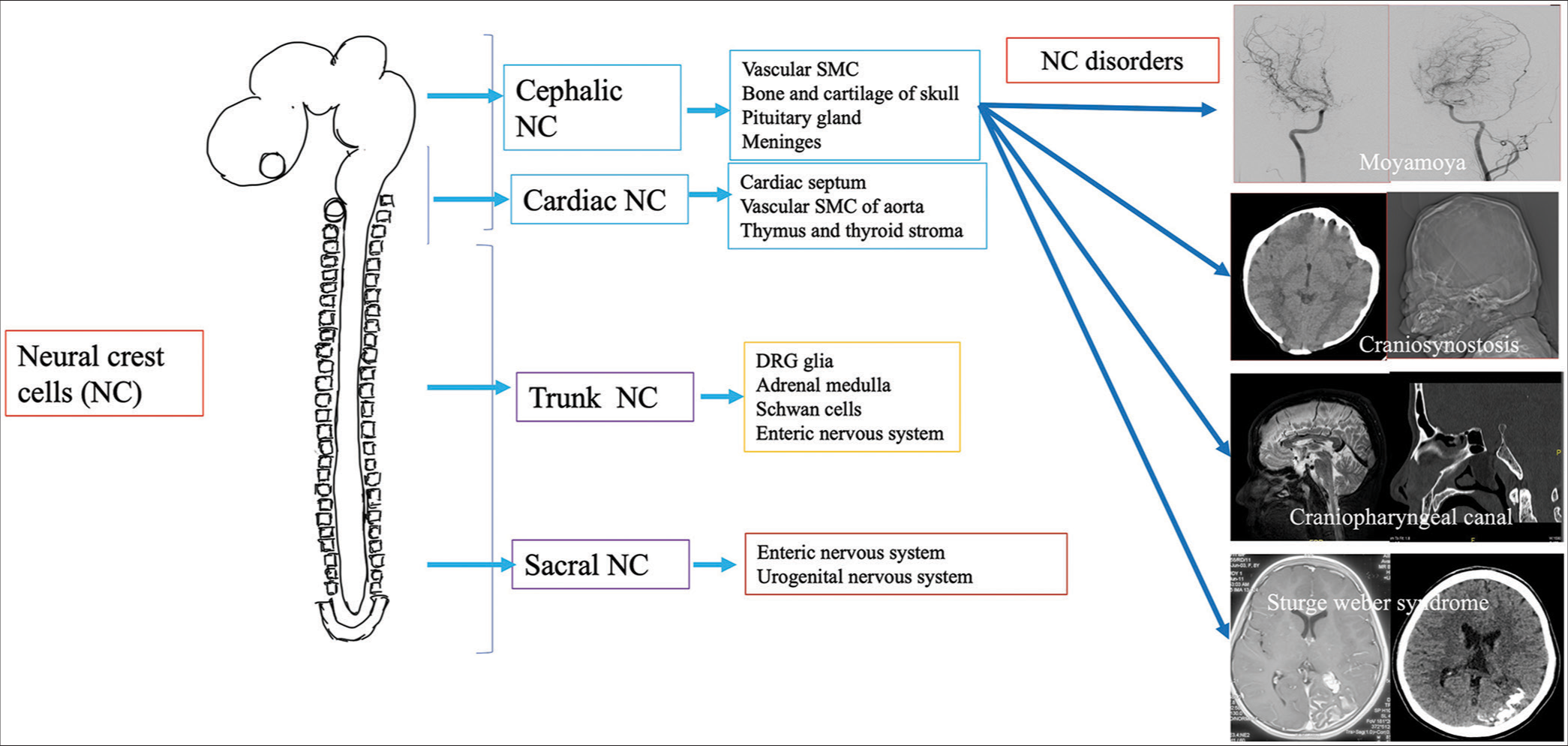
Neural crest cells (NCCs) are transient structures in the fetal life in vertebrates, sharing a common developmental origin for diverse diseases. It develops at the junctional site of the surface and neural ectoderm in the late gastrulation and early neurulation period.[1] The formation of the neural crest (NC) is a complex interplay between various embryonic structures, and a sequence of molecular signals and chemical gradients, especially bone morphogenetic protein.[2] After undergoing epithelio-mesenchymal (EMT), delamination NCCs occur and further migration into various locations of the entire body, producing cell lineage of melanocytes, bones, and cartilage of the cranium, cells of the enteric and peripheral nervous system. The types of NCCs are cranial, vagal, trunk, and sacral, corresponding to various locations in the embryonal neuraxis [Figure 1]. The migration of NCCs is presumed to be filopodial extension and retraction and usually follows a specified pattern according to the final cell type present in the body.[3] For example, neurogenic precursors migrate in the medial pathway below the dermomyotome to make their endpoint, which forms the sympathetic ganglia, dorsal root ganglia, and enteric neural system. In contrast, melanocyte precursor cells choose to migrate in a lateral pathway and are finally located in the skin.[4] In the conventional germ layer model, endodermal derivatives are derived from endodermal cells and mesodermal derivatives from mesodermal cells. Although NCCs are an ectodermal derivative, they can be differentiated into mesodermal or endodermal tissues due to EMT[5] and are thus considered the fourth germ layer.[6] Multiple tissues originate from NCCs and are causative factors in many head and neck pathologies. In the present study, we briefly describe the NC derivatives and 17 cases of this rare group of entities (cephalic NC diseases) who presented to our department in the past 3 years.
Export to PPT
NC DERIVATIVESNCCs constitute to produce various types of tissues and cells, including cartilage and bone of the face; cells of autonomic, enteric, and peripheral nervous systems; and various organs within the body such as the thyroid gland, heart, eye, and adrenal medulla.[3]
NEUROCRISTOPATHIES (NCPS) CLASSIFICATIONA wide variety of diseases (>50 diseases) are associated with disorders of NCCs, which were discovered mostly after the NC theory by Robert Bolande. As understanding of the NCCs progresses, many unrelated diseases are classified under NCP under a single umbrella-like involvement of endocrine, craniofacial, cardiac, immunologic, dermatologic, and gastrointestinal, neoplastic conditions. NCPs can be classified into dysgenetic form due to defective morphogenesis, neoplasms derived from NCCs, combined dysgenesis, and neoplastic etiology.[7,8] Some authors describe vascular NCP as a distinct entity.[9] From the neuro-interventional radiologist’s perspective, vascular and cranial forms of Neurocristopathies (NCPs) are essential. Understanding pharyngeal arch derivatives is essential as NCCs migrate into pharyngeal arches before the closure of the neural tube in gestational life, developing into structures of the face and neck.[10]
Cranial NCCs predominantly migrate and are located in the first, and second pharyngeal arches and pouches with >30 diseases derived from the cranial NCC are described in the literature.[11] Common diseases include holoprosencephaly, craniosynostosis, Parry Romberg disease, hemifacial microsomia–Goldenhar syndrome spectrum, and Treacher Collin syndrome which are of neuroradiologic importance.
MATERIAL AND METHODSA retrospective study was conducted at the Neurosciences Centre, All India Institute of Medical Sciences (AIIMS), New Delhi, India. The study encompassed patients diagnosed with neural crest diseases during the period from January 1, 2017, to December 31, 2019. Comprehensive patient clinical information and imaging data were extracted and archived from GE’s Picture Archiving and Communication Software (PACS).
To identify relevant cases, specific keywords such as ‘neural crest cells’ and ‘neurocistopathy’ were employed in the search process. Eligibility criteria were subsequently applied to ensure the inclusion of cases with both imaging records and clinical follow-up information.
Two independent investigators (MKN and KS) conducted a thorough assessment of imaging studies, including computed tomography (CT), magnetic resonance imaging (MRI), and digital subtraction angiography (DSA).
RESULTSIn this three year study duration, seventeen patients were diagnosed with cephalic neural crest disease, revealing distinct manifestations within the cohort. Among the patients, 6 (35.3%) exhibited the vascular form, 5 (29.4%) presented with the dysgenetic form, 4 (23.5%) demonstrated the mixed form, and 2 (11.7%) displayed the neoplastic form. Notably, brain involvement was observed in all patients (100%), manifesting as either vascular, parenchymal, or a combination of both. Skin involvement was identified in 6 (35.3%) patients, while 2 (11.7%) displayed eye involvement, and 1 (5.9%) exhibited facial involvement. Treatment strategies were tailored according to the specific disease manifestations, and the details of these interventions are thoroughly discussed subsequently. In the following section we briefly describe the clinical and radiological findings in these seventeen patients. Additionally, a summary of the salient features of these cases is provided in Table 1 for a clearer understanding of the observed patterns and variations.
Table 1: Shows a summary of important characteristics of the cases included in this series
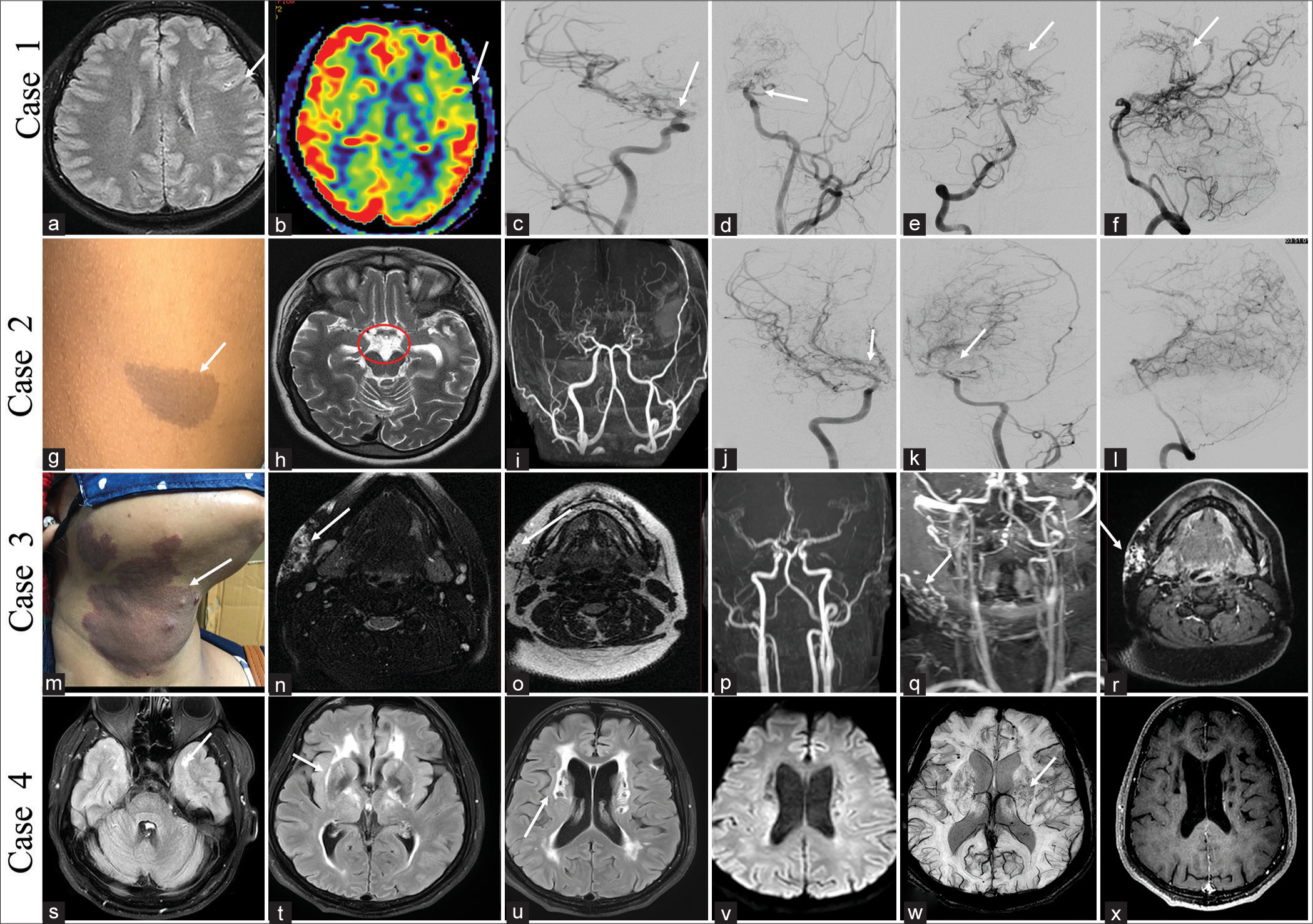
Case with Sl. No. Age, gender Age at presentation Predominant Presenting features Any other Important Distinguishing Clinical feature Other Organ Systems Affected apart from CNS Imaging NCCT MRI Other imaging Histolo-gy if any Genetic Work- upA 12-year-old female child presented with sudden-onset left-side weakness for 1 day, which improved within the next 24 h. There was a history of a similar episode 3 months back, which recovered within 3 days. At the presentation, she had recovered almost fully and was conscious and oriented without any cranial nerve abnormality and normal tone and power and sensory system examination. Magnetic resonance imaging (MRI) brain showed T2/fluid-attenuated inversion recovery hyperintensity in the sulcal space suggestive of Ivy sign, and digital subtraction angiography (DSA) revealed narrowing of both supraclinoid internal carotid arteries (ICAs) and multiple basal and pial collaterals suggestive of Moyamoya disease [Figure 2a-f]. She was started on aspirin and had no further incidence of stroke or transient ischemic attack with 10 months of follow-up duration.
Export to PPT
CASE 2A 10-year-old boy presented with acute right-sided weakness of 3 days duration, which improved completely over 24 h. No history of similar attacks in the past. Examination revealed multiple café-au-lait macules over the trunk and plexiform neurofibroma over the right eyelid. MRI brain showed multiple collaterals in the basal cistern, and a time-of-flight (TOF) image of the circle of Willis (COW) showed narrowing of both supraclinoid ICAs with multiple basal collaterals. DSA showed narrowing of both supraclinoid ICAs and multiple basal and pial collaterals suggestive of Moyamoya syndrome [Figure 2g-l]. He was started on aspirin and one episode of same-sided stroke during 1 year of follow-up. He is planning for external carotid artery (ECA)-middle cerebral artery (MCA) bypass surgery from the neurosurgery side.
CASE 3A 25-year-old female without comorbidities presented with swelling over the right upper neck for 2 years. On examination, a reddish patch was seen over that region since birth which was warm on the touch. The swelling was pulsatile, firm in its consistency, and warm. MRI of the neck showed multiple abnormal vessels with nidus in the right upper side of the neck with supply from the ECA, likely suggestive of arteriovenous malformation (AVM) [Figure 2m-r]. She underwent DSA followed by embolization of the AVM.
CASE 4A 42-year-old male without comorbidities presented with a headache suggestive of migraine for the past 5 years. He also had recurrent transient ischemic attacks in the left MCA territory for 1 year. MRI brain showed white matter changes, predominantly in the anterior temporal pole and external capsule, chronic lacunar infarcts, and microbleeds consistent with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [Figure 2s-x]. He was managed conservatively and was clinically stable at 6 month follow up.
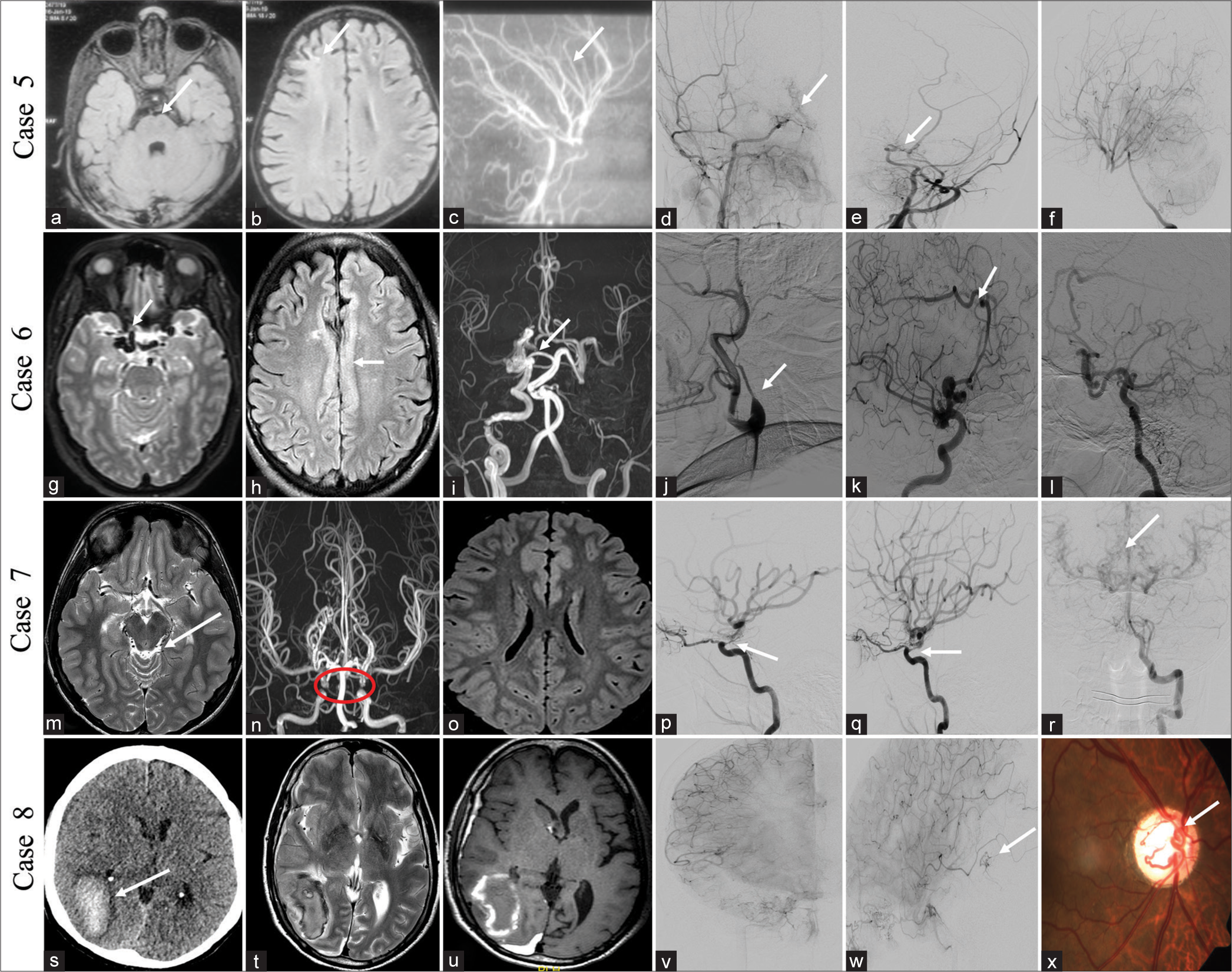
CASE 5A 5-year-old girl presented a history of acute onset-left hemiparesis 3 months back, which partially recovered within 3 days. No similar complaint was seen before. Birth and developmental histories were normal. MRI brain and DSA images showed chronic infarct in the right anterior external watershed region with narrowing of both supraclinoid ICAs without significant collaterals and straightening of vessels, consistent with ACTA2 mutation [Figure 3a-f]. ACTA2 is a non-atherosclerotic cerebral arteriopathy which can involve multisystem smooth muscle due to Arg179His mutations. Angiographic features include ectasia and stenosis of vessels, a straight arterial course, and absence of basal collaterals.
Export to PPT
CASE 6A 48-year-old male without comorbidities presented with intermittent headache, once every month without aura, forgetting names, and irrelevant talk (transient Wernicke aphasia), recovered fully in 4 days. MRI brain findings showed multiple dilated vessels near the right supraclinoid ICA and right pericallosal artery without early draining veins and pachygyria in the underlying cingulate cortex. The DSA images showed a dilated arterial system in the right ICA and right pericallosal artery without abnormal early draining veins suggestive of pure arterial malformations (PAMs). A note was made of the non-visualization of the left ICA from its origin and replaced with an ascending pharyngeal artery [Figure 3g-l]. The patient was managed conservatively and symptomatically improved on 4-months follow-up.
CASE 7A 15-year-old male presented with an intermittent headache for 1 year which was non-pulsatile, frequency of 15 days duration, without aura, and one episode of loss of consciousness (LOC) lasting for 1 h, 5 days back (syncope). Two episodes of slurring of speech and weakness in the right upper limb 10 days prior to presentation, which improved completely over the next 10 hours. His birth and developmental history are unremarkable. MRI image showed multiple abnormal dilated vessels in the quadrigeminal cistern without any signal changes in the brain parenchyma. The TOF image of COW revealed focal non-visualization of the ophthalmic segment of both ICAs. The DSA image showed focal dysgenesis of the ophthalmic segments of both ICAs and multiple pial collaterals reconstituting MCA and ACA branches from the vertebral artery with the diagnosis of segmental dysgenesis of ICAs [Figure 3m-r]. He was managed conservatively with antiplatelets and symptomatic treatment for headache, clinically better with a follow-up duration of 1 year.
CASE 8A 55-year-old male without any previous comorbidities presented with an abrupt onset severe headache associated with one episode of vomiting and loss of consciousness for 15 min. On fundoscopy, an AVM was seen in the retina of the right eye. Non-contrast computed tomography (NCCT) head and MRI brain showed right temporo-occipital lobe subacute hematoma, and DSA showed small AVMs right posterior temporal lobe, fed by a posterior temporal branch of right ICA [Figure 3s-x]. A combination of these findings suggests cerebrofacial arteriovenous metameric syndrome (CAMS) type 2. He underwent conservative management and was doing well till a 2-years follow-up.
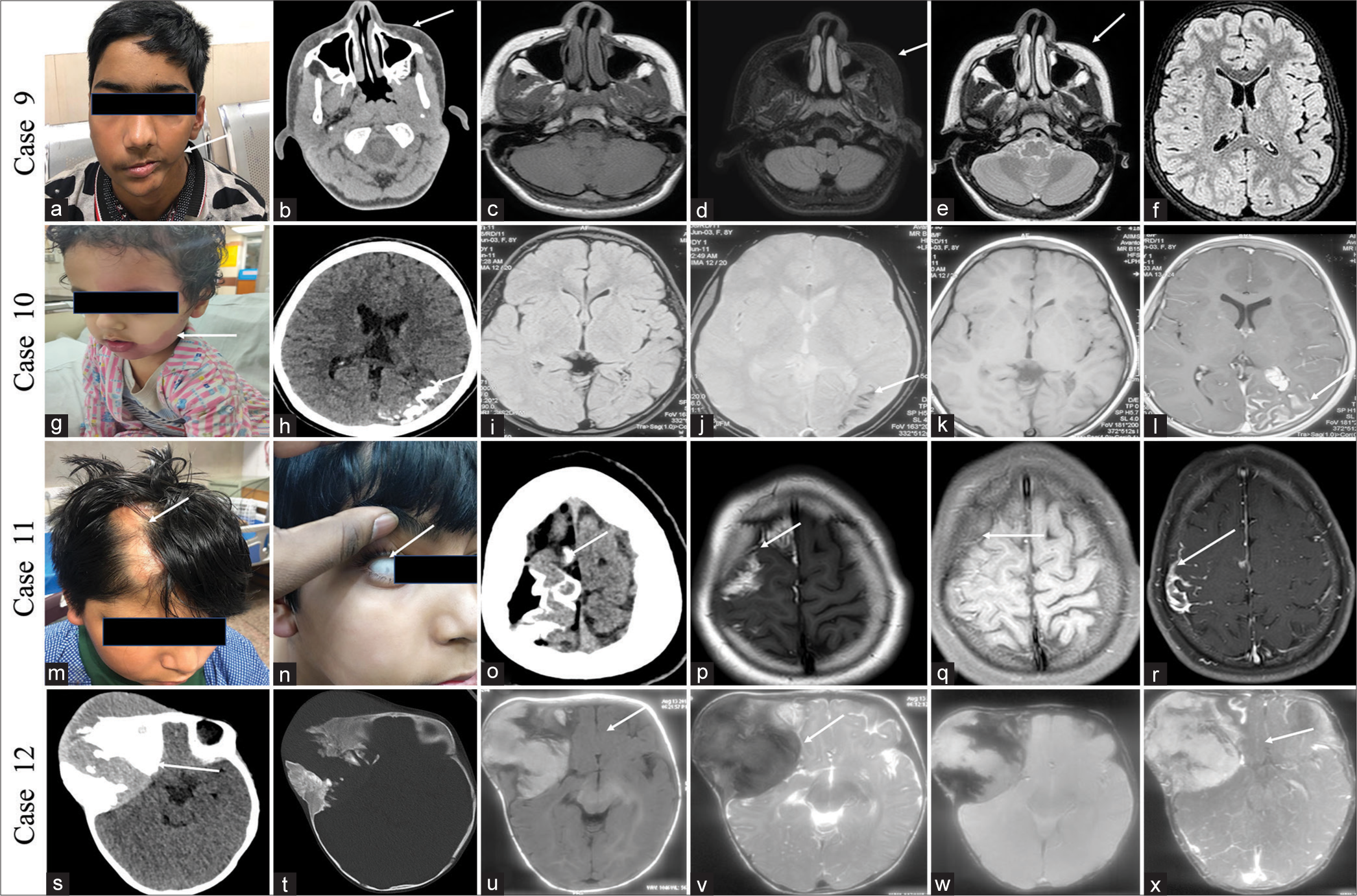
CASE 9A 12-year-old boy presented with progressive left-side facial atrophy since the age of 2 years. His birth and developmental history was normal. No history of seizures or numbness over his face. Clinical examination and MRI showed facial muscle atrophy in the left side without brain parenchymal changes [Figure 4a-f]. The above findings are consistent with Parry-Romberg disease. The patient was managed conservatively and asked for a follow-up.
Export to PPT
CASE 10A 10 year female child presented with 1 episode of generalized tonic-clonic seizure for 1 day. She had suffered a similar episode 3 years ago. Examination revealed a reddish swelling over the left side of the face since birth. The NCCT head showed gyriform calcifications in the left parieto-occipital lobe, and the MRI image showed gyriform calcifications with pial angiomas, consistent with Sturge-Weber syndrome [Figure 4g-l]. She was on two antiepileptics and seizure-free for a 10-month duration.
CASE 11A 13-year-old boy presented with multiple episodes of seizure from 1 year of age and left upper limb weakness for 6 months. He was born of full-term normal vaginal delivery and cried immediately after birth. No history of consanguinity or neurocutaneous manifestations in the family. No history of similar illness in his siblings. Examination revealed patchy hair loss on the right side of the scalp, which has been there for the past 10 years. There was also mild mental retardation. MRI brain showed lipoma with gyriform cortical calcifications in the right superior frontal lobe in both T1, T1 fat saturated, and susceptibility-weighted images. Findings were consistent with the syndrome of encephalocraniocutaneous lipomatosis [Figure 4m-r]. He underwent excision of the lesion, and histopathology showed loss of loss of normal layering with the haphazard arrangement of neurons and fibroadipose tissue. He was seizure-free 2 years after surgery with one antiepileptic.
CASE 12Ten-month-old female child presented with swelling near the right side of the forehead for 9 months, gradually increasing in size. She was born through a full term normal vaginal delivery, cried immediately after birth, and had a normal birth and developmental history. She was having sequential loss of vision in the right eye, followed by the left eye over 6 months. Examination revealed a hard soft-tissue lesion in the right frontotemporal location. Clinical examination and X-ray of the chest revealed no abnormality. Similarly, clinical and ultrasonography examination of the abdomen revealed no abnormality. NCCT head and brain MRI showed a mixed lytic and sclerotic lesion on computed tomography (CT) and T1 hyperintense lesions with post-contrast enhancement in the right frontal and temporal scalp region [Figure 4s-x]. A biopsy of the lesion showed a melanotic neuroectodermal tumor of infancy (MNTI). Seven cycles of neoadjuvant chemotherapy were given to the child (actinomycin D at 0.5 mg, vincristine at 0.7 mg, and cyclophosphamide at 450 mg) over 2 months. The child died 8 months following surgery due to the recurrence of the tumor.
CASE 13A 42-year-old male presented with an intermittent headache for the past 10 years and two episodes of epistaxis within the previous 3 years. He is blind in the right eye from birth with the perception of light and decreased vision in the left eye with hand movement positive in the left eye. No history of trauma was present. On examination, the patient was of short stature. His blood cortisol level was low (4.6 μg/dL), and his thyroid-stimulating hormone level was high (7.9 mIU/L). NCCT scan of the brain showed a defect in the floor of the sella with persistent craniopharyngeal canal (CPC) with the right eye phthisis bulbi and left eye choroidal calcification. MRI of the brain showed transsellar encephalocele through persistent CPC, communicating into the nasopharynx, with lipoma near the nasopharynx (type 3c CPC) and thinning of both optic nerves. Left optic nerve head funneling was seen. TOF COW showed dolichoectatic changes in the vertebrobasilar system without a Moyamoya pattern [Figure 5a-e]. He underwent repair of the encephalocele in the department of neurosurgery.
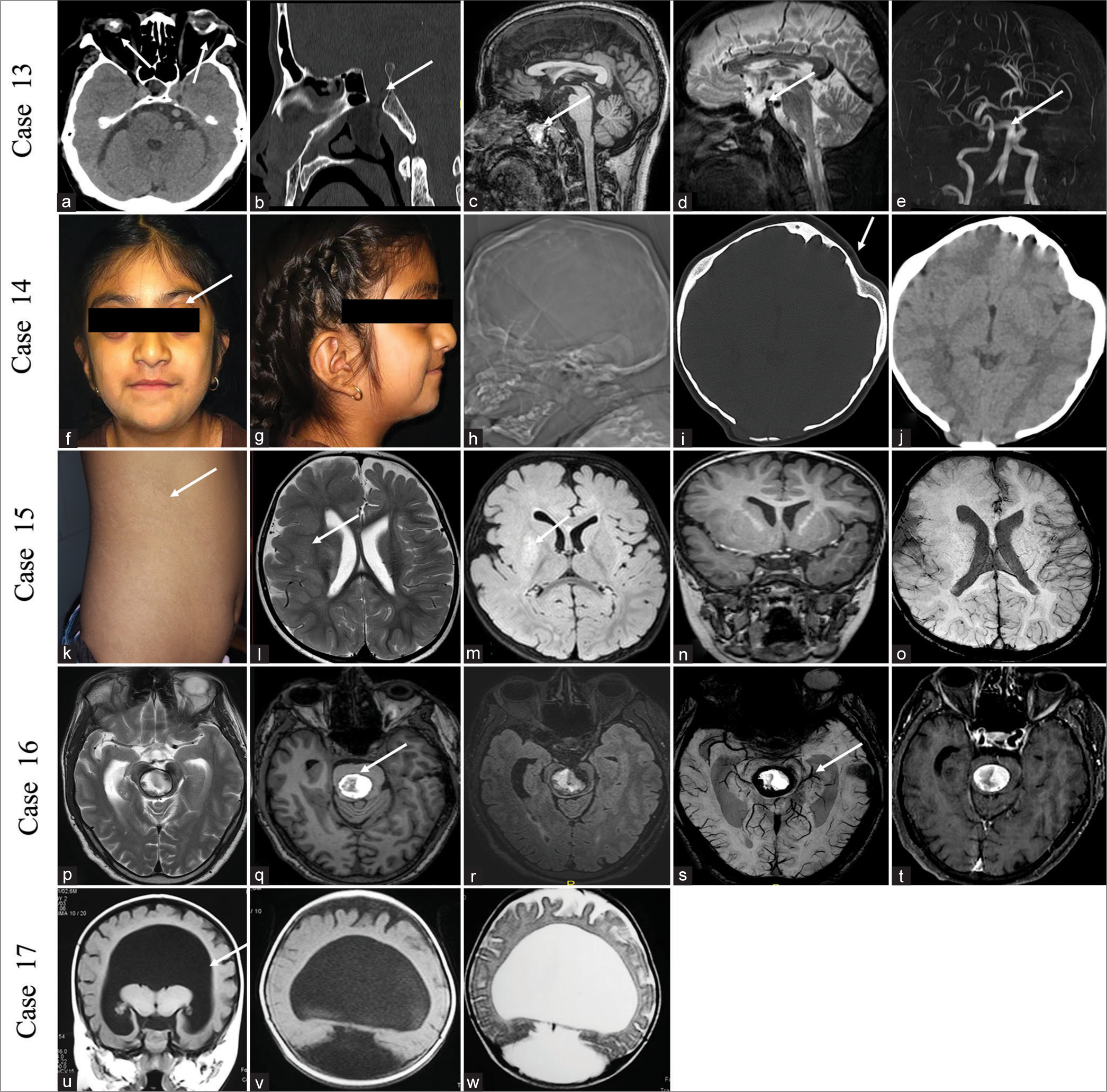
Export to PPT
CASE 14A 12-year-old girl presented with an abnormal skull shape from birth (brachycephaly). Clinical evaluation showed outward protrusion of eyeballs with abnormal skull shape. CT scan showed an abnormal skull shape with reduced antero-posterior (AP) diameter of the skull (brachycephaly) and sutural fusion suggestive of craniosynostosis [Figure 5f-j]. She underwent vault surgery in the department of Neurosurgery.
CASE 15A 10-year-old boy presented with recurrent left-sided focal seizures since 3 months of age. Examination revealed hypopigmented lines over the skin, present along the lines of Blaschko. MRI brain showed a focal abnormal thickened cortex with white matter signal changes in the right frontotemporal lobe. Findings are suggestive of Hypomelanosis of Ito Hypomelanosis of Ito (HI) [Figure 5k-o]. He was managed with antiepileptics and seizure-free for 6 months duration of follow-up.
CASE 16A 41-year-old male without any comorbidities presented with difficulty in walking and progressive weakness in bilateral upper and lower limbs for 1 year. He also complained of one episode of slurring of speech 10 days back, which improved completely within 24 h. MRI brain showed a well-defined T1 hyperintense lesion with peripheral blooming in the central aspect of the pons [Figure 5p-t]. An excisional biopsy of the lesion showed melanocytoma. The patient was followed up for 6 months and showed improvement in power in all four limbs with improvement in his gait.
CASE 17A 6-month-old male child presented with a history of global developmental delay and facial dysmorphism (ocular hypotelorism). MRI image showed non-separation of both cerebral hemispheres with monoventricle and partially fused thalami suggestive of alobar holoprosencephaly [Figure 5u-w].
DISCUSSIONThe NCCs are divided into different types according to their location with their specific derivatives and functions.[3] The cephalic NC develops into the cranial and facial skeleton, including the skull, hypobranchial skeleton, jaws (both upper and lower), adenohypophysis, carotid body, and eye tissues. Cephalic NCCs can produce pericytes and smooth muscle cells (SMCs) of cerebral and facial arteries in the forebrain.[12] However, cardiac NC is located within cephalic NC, which extends from the otic region to the 3rd somite, forming tunica media of the great vessels, coronary arteries, and the aorticopulmonary septum.[12] Thus, abnormal cephalic and cardiac NCCs may result in vascular lesions in Moyamoya disease, PHACE syndrome, and ACTA2 mutation syndrome.
During embryonic development, ICA divides into caudal and rostral segments at the posterior communicating artery (PCom). The caudal division involves Pcom and the first segment (P1) of the posterior cerebral artery (PCA) and rostral division constituting ICA and its distal branches after Pcom. However, the distal PCA branches (P2-4) develop from distal annexation between P1 PCA and telencephalic segments of the anterior choroidal artery.[13] According to Lasjaunias, the distal basilar artery after the primitive trigeminal artery is considered to be the caudal division of ICA, and hence, the embryological boundary between both posterior and anterior circulations is located between the superior cerebellar artery and PCA. A similar observation was seen in our cases 1, 2, and 5, where anterior circulation was observed with relative sparing of the posterior circulation.
Cephalic NC constitutes tunica media of arteries of the anterior circulation, while the posterior circulation originates from the mesoderm. However, the endothelium is derived from the mesoderm in all vessels within the body, including cerebral vasculature. For this reason, Moyamoya disease, PHACE syndrome, ACTA2 mutation, and preferential involvement of anterior circulation with sparing of the posterior circulation.[14] ICA develops from seven different segments between carotid bifurcation and Pcom based on primitive arteries as a segmental concept by Lasjaunias.[15] Multiple spectrums of segmental pathologies may be regression, agenesis, duplication, fenestration, dysplasia, stenosis, looping, rete formation, and dolichoectasia. In one of our cases (case 7), we have seen segmental dysgenesis of bilateral ICA where abnormal NC migration resulted in segmental lesions in ICA.[13]
Pure arterial malformations (PAMs) are arterial malformations described as abnormally dilated and tortuous vessels producing a coil-like configuration without having an early draining vein. The most accepted mechanism for PAM is due to arterial dysplasia caused by a congenital defect or an insult by Brinjikji et al.[16] PAM can be linked with focal cortical dysplasia, corpus callosal dysgenesis, and hemimegalencephaly as arterial development is believed to be influenced by underlying brain development.[17]
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) develops due to an abnormal mutation in the NOTCH3 gene, which resulted in vascular smooth muscle cells (VSMCs), and pericyte degeneration.[18] VSMC is presumed to be of NCC origin, and the Notch gene is responsible for NC differentiation during the cardiovascular system and smooth muscle development.[19]
CAMS is characterized by unilateral cerebral and facial involvement with the medial, lateral prosencephalic group, and lateral rhombencephalic groups.[20]
留言 (0)