
記住我
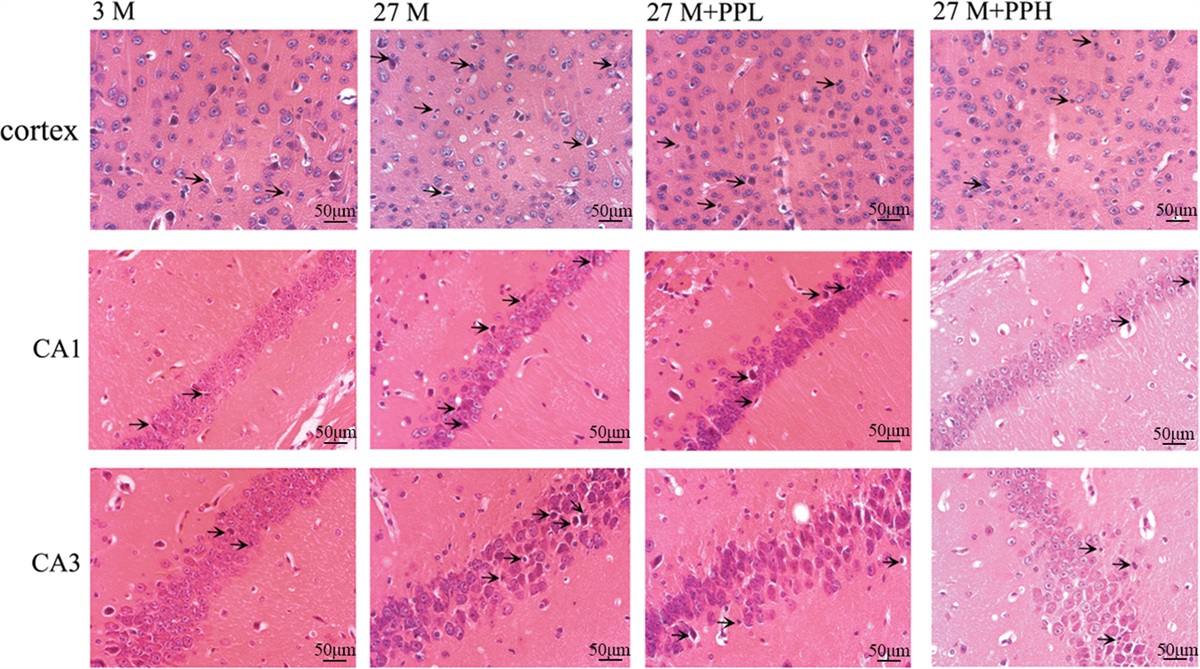


Stroke ranks second to heart disease and cancer as a major factor of disability and mortality globally and imposes a considerable social and economic burden on patients [1]. In recent years, the development of mRNA microarray and transcriptomic analysis has enabled to identify disease biomarkers [2]. Accumulating evidence indicates that a substantial influx of immune cells is crucial during the initial stages of ischemic stroke. The infiltration of resident microglia and peripheral immune cells contributes to the death of neurons and disruption of the blood-brain barrier (BBB) through the release of inflammatory molecules [3]. Hence, understanding the mechanisms through which cuproptosis-related genes (CRGs) influence immune infiltration in ischemic stroke could provide novel ideas to reduce the pathological damage following ischemic stroke.
Copper, a trace element, is required for the proper functioning of the cardiovascular, nervous, blood, and other body systems [4]. Dysregulated copper homeostasis could trigger cytotoxic responses; furthermore, aberrant intracellular copper levels are associated with the occurrence and progression of several diseases. For instance, copper levels are enhanced in tumor tissues and serum of patients with lung, breast [5], and prostate cancers [6]. Dysregulated copper homeostasis is also closely related to neurodegenerative diseases [7]. Cuproptosis, a copper-induced programmed cell death form, occurs when excessive levels of copper ions promote aggregation of lipoylated mitochondrial enzymes, eventually triggering mitochondrial stress and cell death [8,9]. A recent study reported that a copper-dependent antineoplastic agent inhibits the growth of malignant hematopoietic cells [10]. Intracellular copper mainly exists as a cofactor of mitochondrial cytochrome c oxidase and superoxide dismutase and participates in iron utilization, redox balance, oxidative phosphorylation, and cell growth [11]. Iron utilization affects the storage of mitochondrial ferritin that protects nerve cells from damage under stress [12]. Oxidative stress and bioenergetic deficits are also closely related to cerebral ischemia-reperfusion injury [13]. These findings strongly indicate that cuproptosis may function as a pathological factor in stroke occurrence, although its potential regulatory mechanisms remain unclear. Therefore, identifying CRGs clusters involved in stroke could provide novel perspectives regarding the prevention and treatment of stroke.
Here, we screened the differentially expressed CRGs (DECRGs) between stroke patients and healthy individuals and determined two cuproptosis-associated gene clusters in stroke based on eight significant CRGs. These clusters differed in the expression levels of CRGs and immune cell infiltration. Subsequently, cluster-specific differentially expressed genes (DEGs) were identified through weighted gene co-expression network analysis (WGCNA), and gene set variation analysis (GSVA) was performed to elucidate their stroke-related biological functions and pathways. WGCNA has been widely used as a functional interpretation tool in stroke-related bioinformatics research. Based on our findings, we constructed a CRG-based model to predict stroke risk and validated its predictive performance by using an external dataset.
Materials and methods Data collectionThe microarray data were searched in the Gene Expression Omnibus database by using two keywords: ‘ischemic stroke’ and ‘homo sapiens’ [porgn: txid9606]. The ischemic stroke datasets that met the following conditions were selected: (i) based on human peripheral whole blood samples, (ii) included data from patients and healthy subjects, (iii) contained at least more than 20 samples, and (iv) ischemic stroke was diagnosed by MRI or computed tomography. The following three ischemic stroke-related microarray datasets were selected: GSE58294, GSE16561, and GSE22255. We normalized and log2 transformed each expression matrix and then matched it with the platform annotation file. To ensure data accuracy, the average expression value of each gene corresponding to multiple probes was calculated. The R package ‘sva’ was used to remove heterogeneity introduced by different platforms and experimental batches.
Identification of differentially expressed genes associated with cuproptosisBased on a previous study, 19 human CRGs were identified (Supplementary Table 1, Supplemental Digital Content 1, https://links.lww.com/WNR/A721). After normalization and log2 transformation of the original data, the ‘limma’ package in R (version 4.2.1, https://posit.co/products/open-source/rstudio/) was utilized for screening the DECRGs between the ischemic stroke and non-ischemic stroke groups, with cutoff values of |log2Fold Change| > 1 and P < 0.05.
Evaluation of immune cell infiltrationThe immune cell subtypes in the samples were identified using the deconvolute_Xcell function for identifying cell types by determining the relative subsets of RNA transcripts with Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) in the immunedeconv package (version 2.0.4). Immune cell populations in the ischemic stroke (n = 55) and non-ischemic stroke (n = 45) samples were compared using the R software packages ‘ggplot 2’, ‘reshape2’, and ‘ggpubr’.
Relationship of cuproptosis-related genes with infiltrated immune cellsThe correlation of CRGs with the relative percentage of infiltrating immune cells was determined by calculating Spearman’s correlation coefficient (CC) using the ‘tidyverse’, ‘limma’, and ‘reshape2’ packages in R software. Statistical significance was considered based on P < 0.05. The ‘corrplot’ and ‘ggplot2’ packages in R were utilized to display the results and generate heatmaps, respectively.
Analysis of consensus cluster cuproptosis-related genesBased on the eight DECRGs, the 59 ischemic stroke samples in the training set were assigned to different clusters by using the consensus clustering method of the ‘ConsensusCluster-Plus’ package in R software and the k-means algorithm with 1000 iterations. We selected the maximum number of subtype k as 9, and the optimal number of clusters was determined from the consensus matrix, cumulative distribution function (CDF) curve, and consistent clustering score (>0.7). Two clusters were eventually obtained.
Gene set variation analysisWe used GSVA to evaluate variations in the enriched gene sets among the different CRGs. For identifying biological functions and differentially expressed pathways, GSVA scores among CRG clusters were compared using the ‘limma’ package through the ‘GSVA’ and ‘GSEABase’ packages in R software and the ‘c2.cp.kegg.symbols.gmt’ and ‘c5.go.symbols.gmt’ files obtained from the molecular signature database.
Weighted gene co-expression network analysisWe used the ‘WGCNA’ package in R software for screening the ischemic stroke-related pathways and genes in the dataset obtained from GSVA. The top 25% genes showing the largest fluctuations were inputted into the subsequent WGCNA to obtain accurate results. We selected the optimal soft power for constructing a weighted adjacency matrix, which was converted to a topological overlap matrix (TOM). The TOM dissimilarity measure (1 − TOM) with the hierarchical clustering tree algorithm was used to obtain the modules, with the minimum module size of 100. Subsequently, the pathways/genes closely related to ischemic stroke were screened. Pearson’s CC was calculated to determine the correlation of each module with the feature, and the parameter module significance was used to express the relationship between the disease state and the module.
Construction of the predictive model using machine learning methodsFour machine learning predictive models, including random forest, support vector machine (SVM), generalized linear model (GLM), and eXtreme gradient boosting (XGB), were established for the two CRG clusters by utilizing the ‘caret’ package in R software. The recursive feature elimination (RFE) algorithm is used for screening the optimal genes from the training cohort to avoid overfitting, and the most powerful gene set is then identified by SVM-RFE. XGB, an ensemble of boosted trees with gradient boosting, enables a more detailed analysis and comparison of classification error and model complexity. The ‘DALEX’, ‘kernlab’, and ‘xgboost’ packages were used to interpret these models and to visualize their residual distribution as well as important features. Receiver operating characteristic (ROC) curves were generated, and the area under the curve (AUC) value was calculated for screening the model with the best predictive performance by using the ‘pROC’ package in R. The diagnostic significance of the selected model was validated in GSE58294 by ROC analysis.
Construction and validation of the nomogram modelA nomogram consisting of the top predictive CRGs was developed using the ‘rms’ package in R for predicting ischemic stroke risk. Each predictor was denoted a score. We summed the scores of all predictors to determine the risk score. The nomogram model’s predictive power was determined with decision curve analysis (DCA) and calibration curves.
External dataset validation analysisThe predictive model’s ability to differentiate ischemic stroke patients from non-ischemic stroke controls was validated in GSE58294 by ROC analysis using the ‘pROC’ package. The infiltration levels of 28 immune cell populations were compared between the two groups by using the ‘GSVA’, ‘GSEABase’, ‘tidyverse’, ‘ggpubr’, and ‘ggExtra’ packages in R software. We then analyzed the correlation of the CRGs with immune cell infiltration.
To further assess the potential mechanism through which CRGs are involved in ischemic stroke occurrence and development in the model, the expression levels of these genes were compared between the ischemic stroke and non-ischemic stroke groups by using the ‘limma’ package, and their correlations with clinical features were analyzed with the ‘ggpubr’, ‘ggplot2’, and ‘ggExtra’ packages in R software. Finally, the ‘pROC’ package was utilized to estimate the predictive power of all CRGs in the model. P < 0.05 was considered to indicate statistical significance.
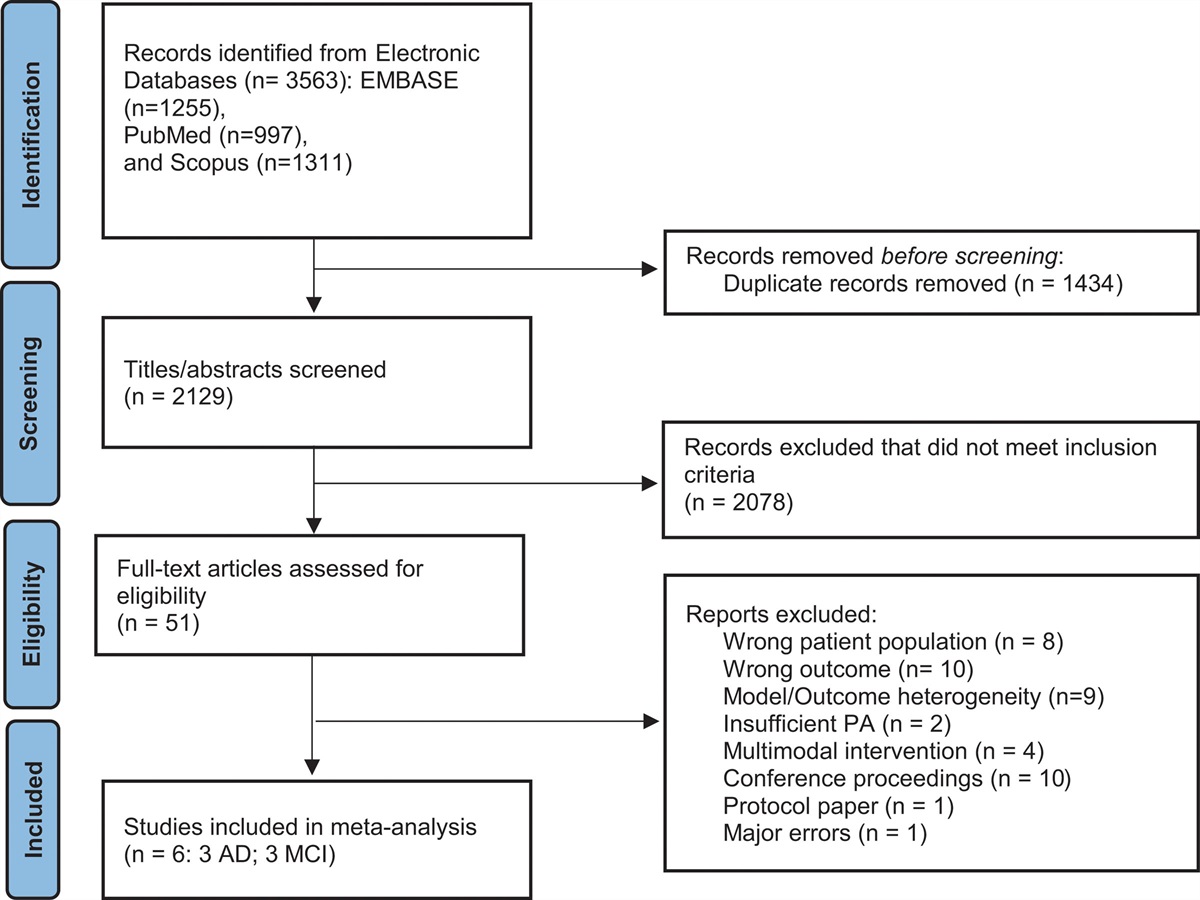
Results Data preprocessingThe GSE16561, GSE22255, and GSE58294 datasets were retrieved on 26 August 2022. The training group (GSE22255 and GSE16561, Table 1) included 59 ischemic stroke patients and 44 non-ischemic stroke controls, and the validation group (GSE58294) included 69 ischemic stroke patients and 23 non-ischemic stroke controls. According to the corresponding platform annotation information obtained from the GSE58294 (GPL570 platform), GSE22255 (GPL570 platform), and GSE16561 (GPL6883 platform) datasets, we converted the probes into gene symbols by using Perl language. Finally, we obtained expression files of 19 CRGs in the ischemic stroke and non-ischemic stroke samples. Figure 1 shows the study flowchart.
Table 1 - Clinical characters of the training data set Total sample, N (%) Fig. 1:
Fig. 1: Overview of the study flow chart.
Identification of differentially expressed cuproptosis-related genesWe identified eight DECRGs, including NFE2-like bZIP transcription factor 2 (NFE2L2), NLR family pyrin domain containing 3 (NLRP3), lipoyltransferase 1 (LIPT1), pyruvate dehydrogenase E1 subunit alpha 1 (PDHA1), metal response element binding transcription factor 1 (MTF1), pyruvate dehydrogenase E1 subunit beta (PDHB), dihydrolipoamide branched chain transacylase E2 (DBT), and glutaminase (GLS) (Fig. 2a and d), of which five genes showed significant downregulation and three genes showed upregulation in the ischemic stroke group (Fig. 2a). To obtain insights into the evolutionary history of the CRGs, we analyzed their chromosomal location by using the CIRCOS genome visualization tool (Fig. 2b). We also analyzed the correlation between these CRGs to elucidate their potential roles in the development of ischemic stroke. As shown in Fig. 2c, NLRP3 and NFE2L2 showed a strong synergistic effect, whereas LIPT1 had the opposite effect.
 Fig. 2:
Fig. 2: Identification of dysregulated CRGs in ischemic stroke and analysis of the difference of immune infiltration between the control group and ischemic stroke group. (a) Boxplots of differential expression of eight CRGs between ischemic stroke and non-ischemic stroke controls. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns means no significance. (b) The location of 19 CRGs on chromosomes. (c) Correlation analysis of eight differentially expressed CRGs. Blue and red represent positive and negative correlations, respectively. The correlation coefficient is represented by the area of the pie chart. (d) Heatmap of expression profiles of eight CRGs. (e) Correlation analysis between eight differentially expressed CRGs and infiltrating immune cells. (f) Analysis of differences in immune infiltration between ischemic stroke and non-ischemic stroke controls. *P < 0.05, ***P < 0.001, ****P < 0.0001, ns, NS. (g) Relative abundance of 22 infiltrating immune cells between ischemic stroke and non-ischemic stroke controls. CRGs, cuproptosis-related genes.
Immune landscape analysisTo confirm the influence of immune cell infiltration on stroke, we compared the immune landscapes of ischemic stroke and non-ischemic stroke samples by using CIBERSORT. The two groups exhibited considerable variations in the infiltrating immune cell type percentage (Fig. 2f and g). The ischemic stroke patients showed a higher infiltration of gamma delta (γδ) T cells, activated dendritic cells, M0 macrophages, and monocytes. We also analyzed whether the infiltrating immune cells were associated with CRGs and noted a positive correlation of activated mast cells with NLRP3 and NFE2L2, but a negative correlation with GLS and DBT (Fig. 2e). M0 macrophages, activated natural killer cells, memory B cells, and M2 macrophages were closely associated with the CRGs (Fig. 2e).
Molecular subtypes of ischemic stroke based on cuproptosis-related genesWe used consensus clustering of the gene expression data to identify new disease subtypes and improve classification for treatment. The CDF and CDF delta area curves revealed relatively stable clustering results for two clusters. Furthermore, at k = 2, each subtype showed a consistency score of >0.7. According to principal component analysis, significant differences were noted between cluster 1 and cluster 2, thus indicating the potential of CRG in stroke diagnosis. NFE2L2 and NLRP3 were upregulated in cluster 1, and LIPT1, DBT, and GLS showed significant upregulation in cluster 2 (P < 0.05).
Immune cell infiltration characteristics between cuproptosis clustersWe further analyzed immune cell infiltration levels in the two clusters and found significant differences in their immune landscapes. Cluster 1 showed more memory B cells, activated dendritic cells, and activated mast cells, while cluster 2 was relatively richer in γδ T cells, resting dendritic cells, and M2 macrophages.
Gene module screening and construction of the co-expression networkFor identifying critical gene modules related to ischemic stroke, we utilized WGCNA to establish co-expression networks and modules in ischemic stroke patients and non-ischemic stroke controls. First, we selected 25% genes showing the highest variance in the GSE16561 and GSE22255 datasets; furthermore, we set the soft threshold power to 9 when constructing a scale-free co-expression network. Next, by using the dynamic cutting algorithm, 9 co-expression modules of different colors were obtained, among which the blue-green module that included 1226 genes showed the strongest correlation with ischemic stroke. A positive correlation was noted between the turquoise module and the related genes. The critical gene modules associated with cuproptosis clusters were constructed using parameters β = 7 and R2 = 0.9; the strongest positive correlation was observed between the yellow module and cluster 1.
Gene set variation analysisWe screened the modules most relevant to clinical characteristics and copper apoptosis clusters through WGCNA and obtained 39 key genes after cross-analysis. Further analysis showed that in cluster 2, Nod-like receptor (NLR) signaling pathway, tight junction, viral myocarditis, and gonadotropin-releasing hormone (GnRH) signaling pathway were upregulated, while functional enrichment analysis showed that cluster 2 was associated with nuclear retinoic acid positive regulation of folded protein responses, BCL2 family protein complexes, and receptor catabolic processes.
Construction and assessment of machine learning modelsTo find subtype-specific CRGs with a high diagnostic significance, four validated machine learning models were constructed using the differential expression data between ischemic stroke and non-ischemic stroke samples. XGB and SVM models exhibited lower residuals. The top 10 significant feature genes of each model were ranked, and ROC curves were generated through five-fold cross-validation to evaluate the four machine learning algorithms’ performance in the validation set. The XGB model exhibited the highest AUC value [AUC values for XGB, random forest, SVM, and GLM = 0.923, 0.914, 0.891, and 0.692, respectively], thus indicating that it can accurately distinguish patients from different clusters. Furthermore, the top five predictive genes in the XGB model were BCL2 interacting protein 1, DEAD-box helicase 18 (DDX18), zinc finger protein 512, aquaporin 9 (AQP9), and intercellular adhesion molecule 2 (ICAM2) (Table 2).
Table 2 - Five genes XGB model identified based on machine learning models Gene name Permutation Dropout_loss Label BNIP1 0 0.340 XGB DDX18 0 0.349 XGB ZNF512 0 0.358 XGB AQP9 0 0.366 XGB ICAM2 0 0.375 XGBXGB, eXtreme gradient boosting.
We further evaluated the predictive performance of the XGB model by estimating stroke risk through a nomogram (Fig. 3d) and verified its high accuracy with calibration curves and DCA (Fig. 3a and b). In the external GSE58294 dataset, we validated the five-gene prediction model. Based on the ROC curve, the model had a high prediction accuracy (AUC = 0.921, see Fig. 3c). Therefore, this five-CRG prediction model can effectively distinguish ischemic stroke patients from healthy individuals and contribute to clinical decision-making.
 Fig. 3:
Fig. 3: Validation of the five-gene-based XGB model. (a and b) Construction of calibration curve. (c) ROC analysis of the five-gene-based XGB model based on five-fold cross-validation in GSE58294. (d) Construction of a nomogram for predicting the risk of ischemic stroke clusters based on the five-gene-based XGB model. XGB, eXtreme gradient boosting.
The role of cuproptosis-related genes in ischemic strokeThe expression of the five predicted genes in ischemic stroke and non-ischemic stroke samples was estimated. We found that AQP9 was upregulated in ischemic stroke samples, while DDX18 and ICAM2 were downregulated (Fig. 4a–c). ICAM2 was also negatively correlated with age, and significant differences were observed in immune cell infiltration between ischemic stroke and non-ischemic stroke samples (Fig. 4d and e). AQP9 was positively correlated with eosinophils, Th17, and Th2, but negatively correlated with central memory CD4 T cells and activated CD8 T cells. ICAM2 showed a positive association with effector memory CD4 T cells but a negative association with eosinophils, mast cells, and Th2 cells (Fig. 4f). In the disease model, the AUC values of these five genes exceeded 0.6. In particular, the AUC of ICAM2 was 0.814, thus suggesting its critical involvement in ischemic stroke development.
 Fig. 4:
Fig. 4: ssGSEA analysis and analysis of the relationship of five genes with immune infiltrating cells and their differential expression in ischemic stroke and non-ischemic stroke. (a–c) Analysis of differential expression of five genes in ischemic stroke and non-ischemic stroke. (d) Correlation analysis between clinical traits and five genes. (e) ssGSEA analysis. (f) Correlation analysis between five genes and immune infiltrating cells. ssGSEA, single-sample gene set enrichment analysis.
DiscussionCuproptosis, a recently discovered copper-dependent cell death program, shows a close association with the occurrence and development of various diseases [14,15]. Recent studies have also underscored the relevance of copper homeostasis in immune cell infiltration [16,17]. According to Choi et al. [18], clodoxoline, a commonly used copper chelator, effectively reduces immune cell infiltration, specifically that of CD8+ and CD4+ T cells, which are responsible for causing encephalitis. After acute stroke, various immune cells enter the brain tissue in an orderly manner. Peripheral immune cells such as bone marrow dendritic cells, neutrophils, and macrophages/monocytes, appear within 1 day following stroke, and subsequently, the number of T and B lymphocytes gradually increases. Inflammatory cells and the immune system show critical involvement in stroke development. Furthermore, targeted intervention through immuno-inflammatory therapies and cuproptosis-related biomarkers may provide new diagnostic and treatment options for stroke.
Among the eight DEGs screened here, NLRP3, NFE2L2, and MTF1 exhibited upregulation in ischemic stroke patients, while PDHA1, LIPT1, DBT, GLS, and PDHB displayed upregulation in non-ischemic stroke patients. In terms of immune cell infiltration, compared to non-ischemic stroke patients, ischemic stroke patients showed higher levels of γδ T cells, B cells, monocytes, M0 macrophages, activated dendritic cells, neutrophils, and activated mast cells; this finding was consistent with previous studies [19,20]. In ischemic stroke research, NLRP3 and its downstream proinflammatory cytokines have been extensively studied at an early stage [21,22]. Studies on curcumin have shown that it improves white matter damage in ischemic stroke by NF-κB and NLRP3 inflammasome inhibition [23]. NLRP3 inflammasome activation involves ROS, potassium efflux, mitochondria, and lysosomes. Hypoxia and reoxidation induce NLRP3 activation in BV2 microglia through ROS; this activation is, however, inhibited by the Nrf2/ARE pathway [24]. In cerebral ischemia-reperfusion injury, the NLRP3 inflammasome causes an inflammatory response; increases the levels of C-X-C motif chemokine 1, IL-1, and IL-6; and releases cathepsin B [25]. In this study, NLRP3 correlated positively with memory B cells and activated mast cells and negatively with M2 macrophages and resting dendritic cells. Recent studies indicate that microglia and infiltrating macrophages critically influence the inflammatory response after stroke [26]. However, additional studies should be conducted to comprehend the macrophage-mediated function of NLRP3 in cerebral ischemia.
NFE2L2 (Nrf2) is an important transcription factor that regulates multiple biological pathways, including metal ion balance, heme/iron metabolism, and antioxidant activity. Nrf2 physiologically regulates oxidative stress and inflammation, has critical involvement in neuroprotection, and might be associated with cuproptosis. It stabilizes GPX4 activity and mitochondrial redox balance by controlling copper oxidation-related genes and glutathione regulatory genes. According to previous studies, Nrf2-inducing drugs can significantly reduce neuronal death, brain edema, inflammation, and neurological deficits after cerebral ischemia [27]. Nrf2 participates mainly in maintaining mitochondrial membrane integrity, and together with NLRP3, it is involved in cerebral ischemia-reperfusion injury. Recent research shows that Nrf2 signaling pathway activation can inhibit NLRP3-mediated inflammation and improve cerebral ischemic damage [28]. Nrf2 inhibits NLRP3 inflammasome activity and reduces the expression levels of its components by weakening NF-κB signaling [29].
MTF1 is an important hypoxia-sensitive transcription factor. During distal limb ischemia adaptation after tMCAO, MTF1 is translocated to the nucleus, where it binds to metal response elements and thus upregulates NCX1 [30]. MTF1 exhibited a significant positive correlation with mast cells. Therefore, MTF1 may play a role through mast cells in ischemic stroke. GLS-mediated glutaminolysis and α-ketoglutarate production are critical in regulating immune activation in neuroinflammation [31]. Although only a few studies have investigated the mechanism of CRGs in immune regulation of ischemic stroke, these studies and the present study indicate that CRGs might be critically involved in the immune infiltration of ischemic stroke [32]. Therefore, understanding the regulation of copper homeostasis in immune cells is essential to develop approaches for preventing and treating ischemic stroke.
To further assess potential signaling pathways of CRG in cuproptosis in ischemic stroke, we found two cuproptosis-related clusters through unsupervised cluster analysis. Among them, Cluster 1 included ammonia tRNA biosynthesis and mismatch repair pathways, while Cluster 2 included the NLR and MAPK signaling pathways, with the GnRH signaling pathway as the major component. GnRH signaling is highly correlated with aberrant mitotic and differentiation signaling, abortive cell cycle progression, and eventually neuronal death. Luteinizing hormone/chorionic gonadotropin receptor is critical to maintain the dynamic structure of the BBB and vascular system. The NLR signaling pathway mediates inflammation and apoptosis, and it is the key pathway of ischemic brain injury. We also found that ischemic stroke may be related to the cell cycle and mismatch repair processes, thus suggesting that ischemic stroke might be associated with amino acid metabolism and cell cycle.
Finally, we constructed an ischemic stroke risk prediction model by using a machine learning model of multivariate analysis. These models are increasingly used for disease prediction because they are relatively more reliable. The output of four machine learning classifiers was compared, and XGB was finally used to establish a CRG model comprising five key genes for ischemic stroke risk prediction. We observed that the ICAM2 gene in the model was downregulated in ischemic stroke samples and was negatively correlated with patient age. AQP9 expression was significantly increased in ischemic stroke patients. AQP9, an AQP family member, is critically involved in brain edema development after stroke. In particular, the AQP-9 and AQP-4 levels are increased in the brains of ischemic stroke mice. Furthermore, there is evidence that infarct volume correlates with AQP-4/AQP-9 expression levels [33,34]. ICAM-2 is an intracellular adhesion molecule protein with wide distribution in peripheral blood cells and vascular endothelial cells. It participates in binding of neutrophils to vascular endothelium, cell–cell interactions, and aggregation of platelets and leukocytes; thus, it could function as a biomarker for ischemic stroke diagnosis [35]. The nomogram constructed using the XGB model can predict ischemic stroke risk with high accuracy, thus showing potential clinical application value. In summary, this model showed satisfactory performance in predicting ischemic stroke subtypes and risks.
ConclusionNFE2L2, NLRP3, GLS, LIPT1, and MTF1 could function as predictor genes of cuproptosis and are involved in immune cell infiltration in ischemic stroke. Furthermore, we constructed a predictive model for stroke based on five CRGs to evaluate the subtypes and prognosis of ischemic stroke patients. Our findings generate new perspectives regarding the molecular mechanisms responsible for ischemic stroke progression and heterogeneity and highlight potential research directions for its diagnosis and treatment.
LimitationsFirst, despite meeting the required sample size for the study, the results of the microarray data analysis might be biased due to a relatively small sample size. Second, the data we used are older and potentially heterogeneous. Finally, although we screened for ischemic stroke-related CRGs, the precise mechanisms of action of these genes are yet to be fully elucidated and require further investigation.
AcknowledgementsWe thank Professor Tang for her critical review of our manuscript and her guidance and also our colleagues for their support. We would like to thank Professor Liu for her linguistic assistance during the preparation of this manuscript. C.L. and B.W. conceived and designed the study. B.W., Y.T., and X.L. conducted the study. X.L., Z.W., and Z.G. contributed to data acquisition. Y.T. and D.T. analyzed the data. B.W. and Y.T. interpreted the data. C.L. edited the manuscript draft. D.T. reviewed and edited the manuscript. All authors have read and approved the manuscript. The datasets (GSE22255, GSE16561, and GSE58294) supporting the conclusions of this article can be downloaded from the GEO website (https://www.ncbi.nlm.nih.gov/geo/).
Conflicts of interestThere are no conflicts of interest.
References 1. Owens Johnson CNM, Roth GA, Nichols E, Alam T, Abate D. Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019; 18:439–458. 2. Fan J, Cao S, Chen M, Yao Q, Zhang X, Du S, et al. Investigating the AC079305/DUSP1 axis as oxidative stress-related signatures and immune infiltration characteristics in ischemic stroke. Oxid Med Cell Longevity 2022; 2022:8432352. 3. Ma Y, Yang S, He Q, Zhang D, Chang JJ. The role of immune cells in post-stroke angiogenesis and neuronal remodeling: the known and the unknown. Front Immunol 2021; 12:784098. 4. Baker ZN, Cobine PA, Leary SC. The mitochondrion: a central architect of copper homeostasis. Metallomics 2017; 9:1501–1512. 5. Adeoti M, Oguntola A, Akanni E, Agodirin O, Oyeyemi GJ. Trace elements; copper, zinc and selenium, in breast cancer afflicted female patients in LAUTECH Osogbo, Nigeria. Indian J Cancer 2015; 52:106. 6. Saleh SA, Adly HM, Abdelkhaliq AA, Nassir AM. Serum levels of selenium, zinc, copper, manganese, and iron in prostate cancer patients. Curr Urol 2020; 14:44–49. 7. Gromadzka G, Tarnacka B, Flaga A, Adamczyk A. Copper dyshomeostasis in neurodegenerative diseases—therapeutic implications. Int J Mol Sci 2020; 21:9259. 8. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021; 22:266–282. 9. Kahlson MA, Dixon SJ. Copper-induced cell death. Science 2022; 375:1231–1232. 10. Chen G, Niu C, Yi J, Sun L, Cao H, Fang Y, et al. Novel triapine derivative induces copper-dependent cell death in hematopoietic cancers. J Med Chem 2019; 62:3107–3121. 11. Wang P, Cui Y, Liu Y, Li Z, Bai H, Zhao Y, et al. Mitochondrial ferritin alleviates apoptosis by enhancing mitochondrial bioenergetics and stimulating glucose metabolism in cerebral ischemia reperfusion. Redox Biol 2022; 57:102475. 12. Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B, et al. Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis 2002; 29:376–383. 13. Wang P, Cui Y, Ren Q, Yan B, Zhao Y, Yu P, et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis 2021; 12:1–16. 14. Chen Y, Tang L, Huang W, Zhang Y, Abisola FH, Li L. Identification and validation of a novel cuproptosis-related signature as a prognostic model for lung adenocarcinoma. Front Endocrinol 2022; 13:963220. 15. Lai Y, Lin C, Lin X, Wu L, Zhao Y, Lin F. Identification and immunological characterization of cuproptosis-related molecular clusters in Alzheimer’s disease. Front Aging Neurosci 2022; 14:932676. 16. Yang X, Wang X, Sun X, Xiao M, Fan L, Su Y, et al. Construction of five cuproptosis-related lncRNA signature for predicting prognosis and immune activity in skin cutaneous melanoma. Front Genet 2022; 13:972899. 17. Yuan H, Qin X, Wang J, Yang Q, Fan Y, Xu D. The cuproptosis-associated 13 gene signature as a robust predictor for outcome and response to immune- and targeted-therapies in clear cell renal cell carcinoma. Front Immunol 2022; 13:971142. 18. Choi BY, Jang BG, Kim JH, Seo JN, Wu G, Sohn M, et al. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol Dis 2013; 54:382–391. 19. Palumbo ML, Moroni AD, Quiroga S, Castro MM, Burgueño AL, Genaro AM. Immunomodulation induced by central nervous system-related peptides as a therapeutic strategy for neurodegenerative disorders. Pharmacol Res Perspect 2021; 9:e00795. 20. Muhammad S, Chaudhry SR, Kahlert UD, Niemelä M, Hänggi D. Brain immune interactions-novel emerging options to treat acute ischemic brain injury. Cells 2021; 10:2429. 21. Franke M, Bieber M, Kraft P, Weber AN, Stoll G, Schuhmann MK. The NLRP3 inflammasome drives inflammation in ischemia/reperfusion injury after transient middle cerebral artery occlusion in mice. Brain Behav Immun 2021; 92:221–231. 22. Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK. NLPR3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood–brain barrier integrity in murine stroke. Cell Death Dis 2021; 13:20. 23. Ran Y, Su W, Gao F, Ding Z, Yang S, Ye L, et al. Curcumin ameliorates white matter injury after ischemic stroke by inhibiting microglia/macrophage pyroptosis through NF-κB suppression and NLRP3 inflammasome inhibition. Oxid Med Cell Longev 2021; 2021:1–25. 24. Zhou D, Zhang L, Mao L, Cao J, Gao J. Biological mechanism on SIRT1/NLRP3/IL-18 signaling pathway of acupuncture for treatment of ischemic stroke with center poststroke pain. Comput Intell Neurosci 2022; 2022:8958742. 25. Prakash R, Vyawahare A, Sakla R, Kumari N, Kumar A, Ansari MM, et al. NLRP3 inflammasome-targeting nanomicelles for preventing ischemia-reperfusion-induced inflammatory injury. ACS Nano 2023; 17:8680–8693. 26. Howell JA, Gaouette N, Lopez M, Burke SP, Perkins E, Bidwell GL 3rd. Elastin-like polypeptide delivery of anti-inflammatory peptides to the brain following ischemic stroke. FASEB J. 2023;37(8):e23077. doi: 10.1096/fj.202300309RR. 27. Liu L, Locascio LM, Doré S. Critical role of Nrf2 in experimental ischemic stroke. Front Pharmacol 2019; 10:153. 28. Bai R, Lang Y, Shao J, Deng Y, Refuhati R, Cui L. The role of NLRP3 inflammasome in cerebrovascular diseases pathology and possible therapeutic targets. ASN Neuro 2021; 13:17590914211018100. 29. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019; 19:477–489. 30. Valsecchi V, Laudati G, Cuomo O, Sirabella R, Annunziato L, Pignataro G. The hypoxia sensitive metal transcription factor MTF-1 activates NCX1 brain promoter and participates in remote postconditioning neuroprotection in stroke. Cell Death Dis 2021; 12:423. 31. Wu B, Liu J, Zhao R, Li Y, Peer J, Braun AL, et al. Glutaminase 1 regulates the release of extracellular vesicles during neuroinflammation through key metabolic intermediate alpha-ketoglutarate. J Neuroinflammation 2018; 15:79. 32. Chen W, Chen Y, Wu L, Gao Y, Zhu H, Li Y, et al. Identification of cell death-related biomarkers and immune infiltration in ischemic stroke between male and female patients. Front Immunol 2023; 14:1164742. 33. Halai K, Whiteford J, Ma B, Nourshargh S, Woodfin AJ. ICAM-2 facilitates luminal interactions between neutrophils and endothelial cells in vivo. J Cell Sci 2014; 127:620–629. 34. Tu Q, Yan L, Wang C, Han A, Qin Y, Cui L, et al. Associations between aquaglyceroporin gene polymorphisms and risk of stroke among patients with hypertension. Biomed Res Int 2020; 2020:9358290. 35. Liu W, Yang XZ, Zhang D, He X, Yu Q, Liu X, et al. Differential regulation of the immune system in peripheral blood following ischemic stroke. Biomed Res Int 2022; 2022:2747043.
留言 (0)