
記住我

This phase IIb, randomized, double-blind, placebo-controlled clinical trial was performed in four academic and non-academic ICUs in the Netherlands between March 2021 and March 2022 (Fig. 1, Additional file 1: Figure S1). The trial protocol was approved by the Medical Ethics Committee of the Amsterdam UMC (location VUMC, IRB number NL75871.029.20, approved on 22-01-2021) and has been published [26]. Written informed consent was obtained from the patient’s legal representative. The trial was conducted according to the principles of the World Medical Association’s Declaration of Helsinki.
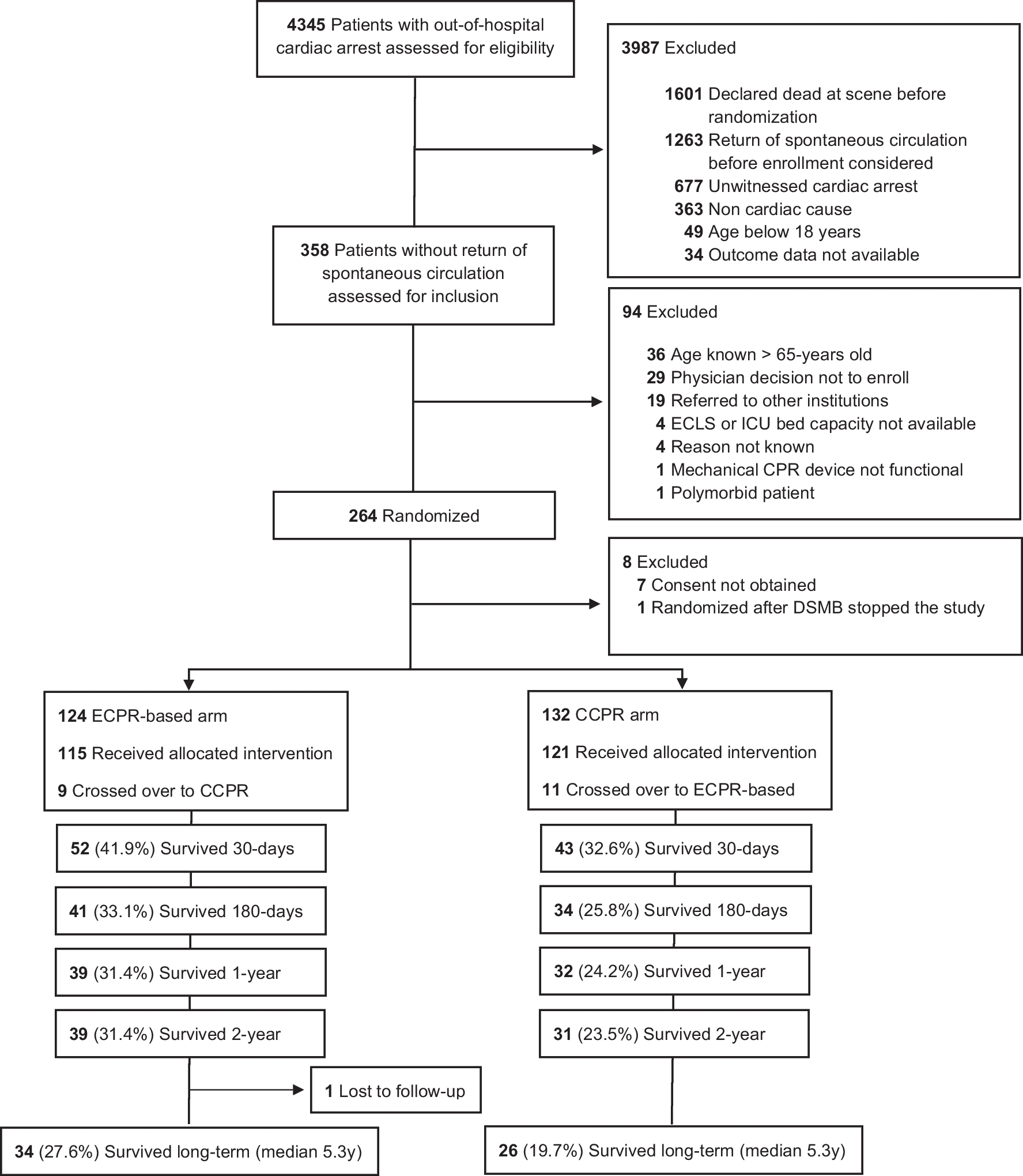
Fig. 1
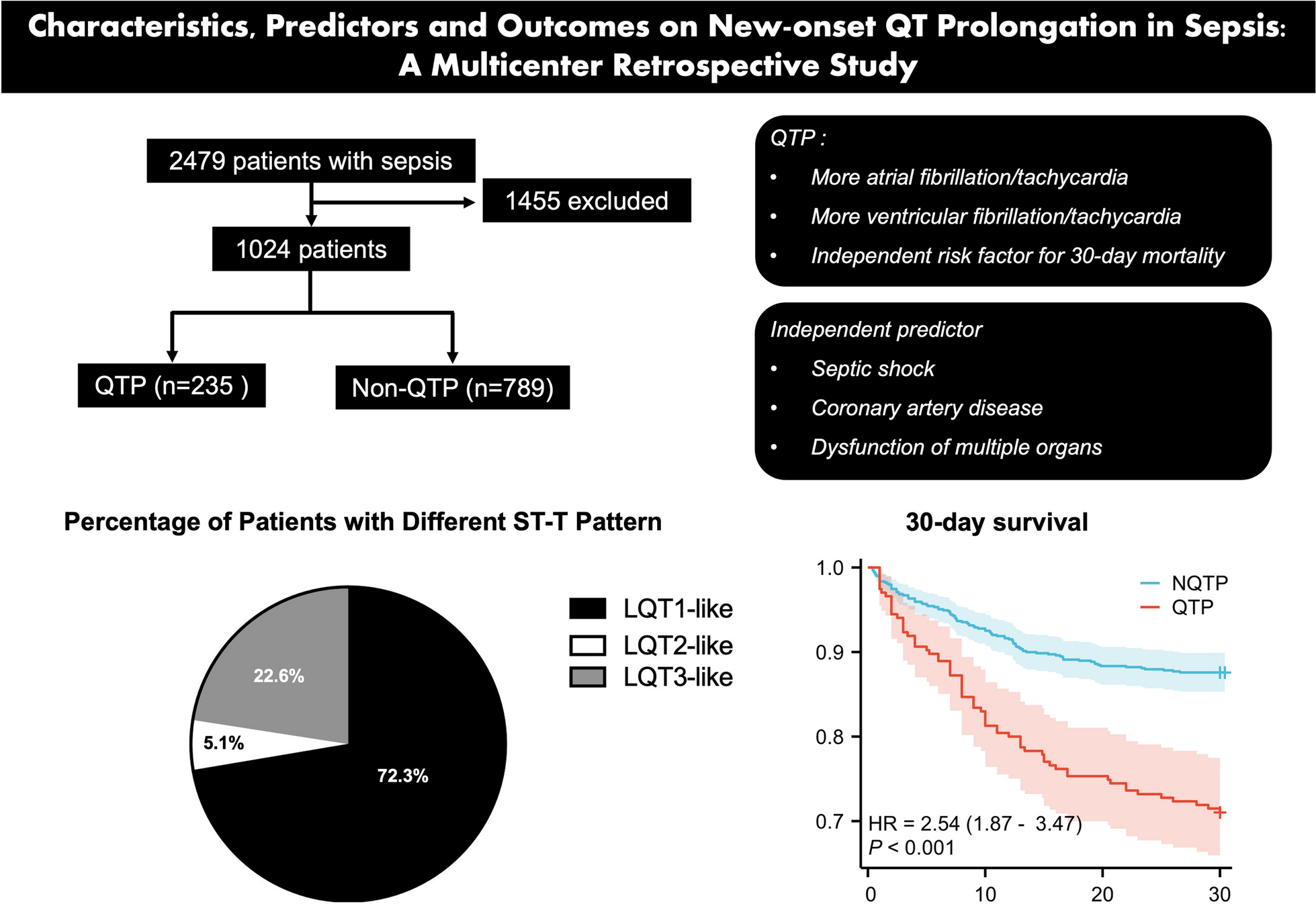
Patient screening and inclusion. Screening and inclusion flowchart. Of the 67 randomized patients, 66 patients were included in the final analysis and 64 patients received at least one dose of the study medication
Patients were eligible if aged ≥ 18 years, intubated for invasive ventilation and had moderate-to-severe ARDS due to COVID-19. ARDS was classified according to Berlin criteria [27]. The patients were included in the trial as soon as possible after intubation and were excluded from participation if the anticipated start of study medication was > 48 h after the start of invasive ventilation. Other exclusion criteria included a history of severe chronic pulmonary disease, ejection fraction of < 40% and participation in another clinical trial. A complete overview of all exclusion criteria can be found in Additional file 1 (p. 2).
Randomization and blindingPatients were randomized 1:1 to receive IV imatinib or placebo for 7 days. Randomization was done in the web-based application Castor electronic data capture using variable block sizes (4–6 patients per block) and stratification per participating center. Allocation of randomization group was only visible to the pharmacy staff preparing the treatment. The patients, clinical staff, investigators and statisticians remained blinded to randomization allocation during the entire study. Blinding was guaranteed by distribution of the study drug in amber-colored syringes and lines to conceal any color differences. The local pharmacies were responsible for the preparation of the study drug and, if necessary, for unblinding.
Study proceduresAt baseline, all patients received a Pulse Contour Cardiac Output (PiCCO; Pulsion Medical Systems, Munich, Germany) catheter. PiCCO catheter placement was performed as part of a deferred consent procedure and replaced the standard care arterial line. Extravascular lung water (EVLW) measurements were performed daily for seven days or until transfer to the ward, as previously described [28, 29]. EVLW was indexed to predicted body weight.
After randomization and PiCCO measurement, the study drug was administered twice daily as a 25 ml, two-hour infusion for seven days or until ICU discharge. The study drug consisted of a 9.6 mg/mL imatinib mesylate solution (Exvastat Ltd, Cambridge, United Kingdom), corresponding to 200 mg imatinib per dose, or placebo. Clinical and ventilation parameters were recorded on days 1,2,4,7,10 and 28. Ventilation parameters were recorded once per hour, and partial oxygen pressure (PaO2) at least once per shift (i.e. every 4 h) or more frequently, if clinically indicated. Ventilation parameters were collected at the time of the lowest ratio of PaO2 to the fraction of inspired oxygen (FiO2). Blood samples were collected for plasma biomarker analyses (Additional file 1: Table S1).
Adverse events (AEs) and serious adverse events (SAEs) were recorded until day 28. Due to the high incidence of adverse events in the ICU, only prespecified events were recorded (Additional file 2, pp. 26–29). Reporting was conducted according to the Council for International Organizations of Medical Sciences (CIOMS) reporting guidelines. Safety was assessed using clinical laboratory tests and electrocardiograms (ECG) at baseline and on days 1,2,4,7 and 10. Details on predefined stopping criteria, are described in the protocol (Additional file 2, pp. 24–25). Patients discharged before day 28 were contacted by telephone on day 28 to evaluate their clinical status.
Prespecified outcomesThe primary endpoint was the change (Δ) in EVLWi between days 1 and 4. We chose this period to capture the effect of imatinib during the exudative phase of ARDS, characterized by vascular leak and alveolar flooding [3, 30, 31]. We hypothesized that this period would be the window-of-opportunity in which imatinib could best exert its vasculoprotective effect.
The following key secondary outcomes were analyzed in hierarchical order: change in PaO2/FiO2 ratio, number of ventilator-free days (VFD), length of ICU, hospital length of stay and 28-day mortality. Other explorative secondary endpoints included the duration of invasive ventilation, change in the pulmonary vascular permeability index (PVPi) and plasma biomarker concentrations (Additional file 1: Table S1), ventilation parameters, Sequential Organ Failure Assessment (SOFA) score and the 9-point World Health Organization (WHO) ordinal scale for clinical improvement (all specified in Additional file 1).
Sample size calculationA sample size of 90 patients was determined to demonstrate a 25% reduction in EVLWi, with 80% power and a type 1 error rate of 0.05. The calculation was based on an anticipated baseline EVLWi of 17 ml/kg with a standard deviation (SD) of 7 ml/kg. This was based on previously performed EVLW measurements in patients with moderate to severe ARDS [32-34]. The expected EVLWi reduction of 25% was based on preclinical data [11].
The study protocol (Additional file 2) allowed for a sample size re-estimation in case of falling recruitment rates. Revision of the power calculation was supported by a lower variation in EVLWi than initially anticipated based on data in non-COVID-19 ARDS (i.e. SD of 4.9 instead of 7.0). Re-estimation was done by updating the assumptions using pre-randomization data from the 66 patients included at the time of re-estimation. The estimated difference between groups (µ1 – µ2) was left unchanged, as we were not able to re-estimate the treatment effect without unblinding. Based on the observed mean EVLWi at n = 66 of 16.5 ml/kg with an SD of 4.9 and assuming an alpha of 5%, 19 patients per allocated group were considered sufficient to detect a 25% reduction in EVLWi between the groups at 80% power. Therefore, recruitment was halted after 67 patients.
Statistical analysisStatistical analyses were performed in the intention-to-treat (ITT) population of 66 randomized patients. An overview of predefined statistical tests is provided in the Statistical Analysis Plan (Additional file 3). In summary, normal distribution was tested using the Shapiro–Wilk test. Baseline imbalances were defined as a difference of ≥ 5% between the placebo and imatinib groups, and baseline imbalances deemed clinically relevant to the primary outcome by consensus were adjusted for in subsequent analyses. Categorical data were expressed as numbers and percentages, and differences between categorical variables were tested using a Chi-square test. Continuous data were expressed as mean ± SD or median with interquartile range [IQR]. Differences between continuous variables were analyzed depending on parametric or non-parametric distribution using a two-tailed t-test, or Mann–Whitney-U-test, respectively.
The primary endpoint was presented as mean ± SD and analyzed using a two-tailed t-test. In addition, an analysis of covariance (ANCOVA) was performed. For the primary endpoint, data imputation was performed in case of missing EVLWi values. In case of missing values on day 1, the data from day 2 was carried backwards. In case of missing values at day 4, the value was imputed by carrying the day 3 value forward, or, in case of no day 3 measurement, carrying the day 5 value backwards. For sensitivity analysis, the primary endpoint was analyzed in the ITT population without data imputation. In addition, a per-protocol analysis of the primary endpoint was performed in all patients missing ≤ 1 study drug dose in the first four study days and who had no missing EVLWi measurements on days 1 and 4.
28-day mortality was analyzed using a Cox proportional hazards model and visualized using a Kaplan–Meier curve. EVLWi, PVPi, ventilation parameters, plasma biomarkers and clinical laboratory outcomes over the first seven days were analyzed using linear mixed effect (LME) models, with time point, randomization group and their interaction term as fixed effects, study ID as random intercept and, in case of baseline imbalances, relevant covariates. For LME analyses, logarithmic transformation was applied to non-normally distributed data.
We performed a posthoc analysis applying the three biomarker-derived subphenotypes identified in the CounterCOVID trial [18]. Using the nnet package [35], a multinomial logistic regression model was trained using data from the CounterCOVID trial with plasma levels of interleukin (IL)-6, tumor necrosis factor receptor 1 (TNFR1), surfactant protein (SP)-D and angiopoietin (Ang)-2 to Ang-1 ratio measured at baseline as predictors of cluster allocation. Based on pre-treatment biomarker data obtained from the InventCOVID patients and using the predict() function in R studio [36], this model was used to classify patients into three subphenotypes: i.e. subphenotype 1 (high IL-6, high TNFR1, low SP-D), subphenotype 2 (low IL-6, low TNFR1, low SP-D) and subphenotype 3 (high IL-6, high TNFR1, high SP-D). LME modelling of ΔEVLWi per treatment day was repeated in the subphenotypes. Statistical analyses were performed using R, version 4.1.3 and RStudio, version 2022.02.1.
留言 (0)