
記住我

The aim of this study is to investigate changes in synaptic plasticity and alertness in patients with RASopathies, namely NS and NF1, after the application of lovastatin (to NS patients) and lamotrigine (to NS and NF1 patients).
DesignThe SynCoRAS study is a monocentre, randomized, double-blind, parallel-group, placebo-controlled, cross-over clinical trial phase IIa. The trial design is shown in Figs. 1 and 2. The clinical trial has been approved before trial commencement by the local ethics committee (vote 461/17 Af dated 13–02-2018, with the last amendment No. 3 dated 12–11-2020) and by the German medical regulatory authorities (Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM)). Prior start of recruitment the clinical trial was registered in ClinicalTrials.gov (https://www.clinicaltrials.gov) as well as in the European Union (EU) Clinical Trials Register (EudraCT-Nr.: 2016–005022-10).
Fig. 1
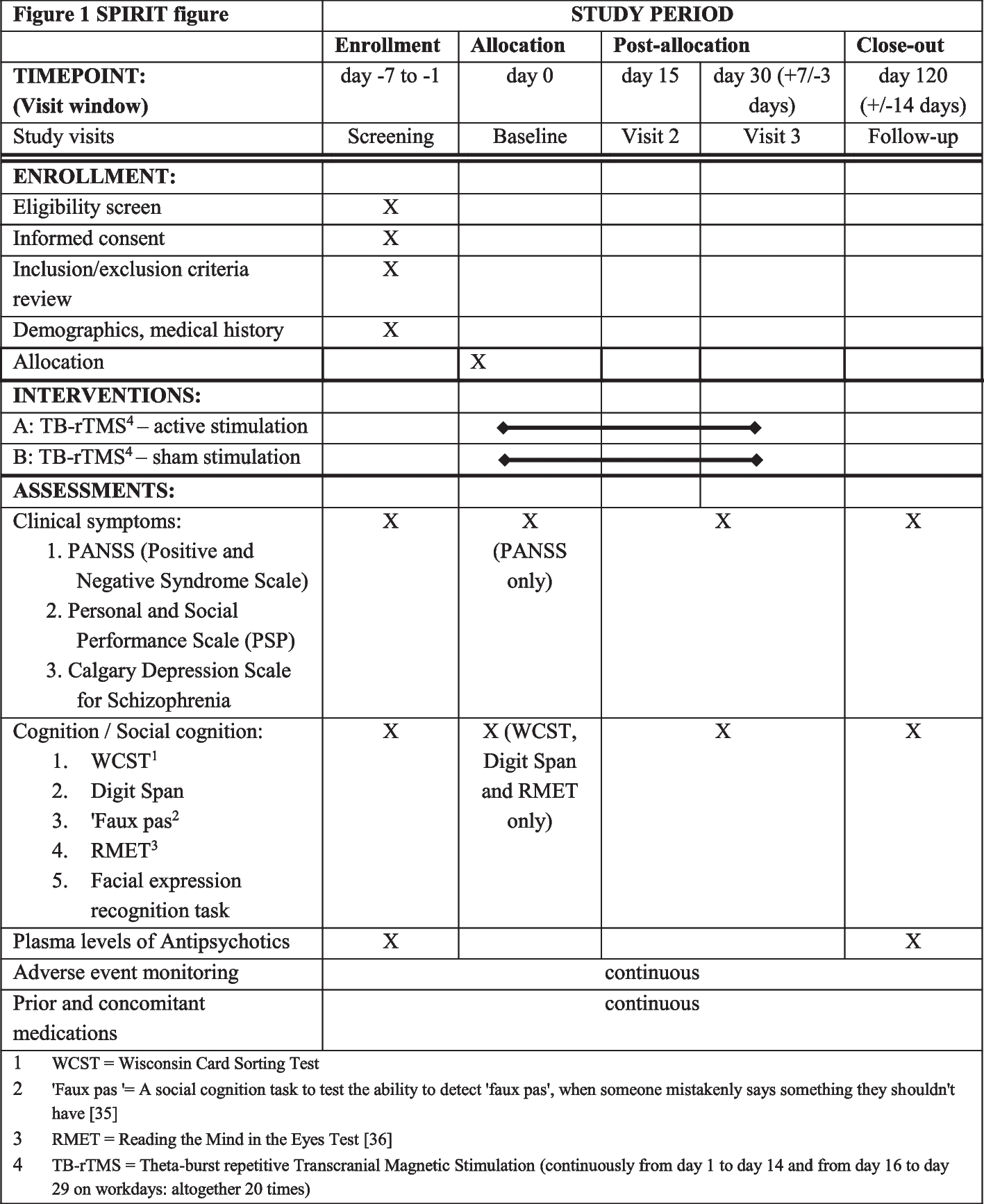
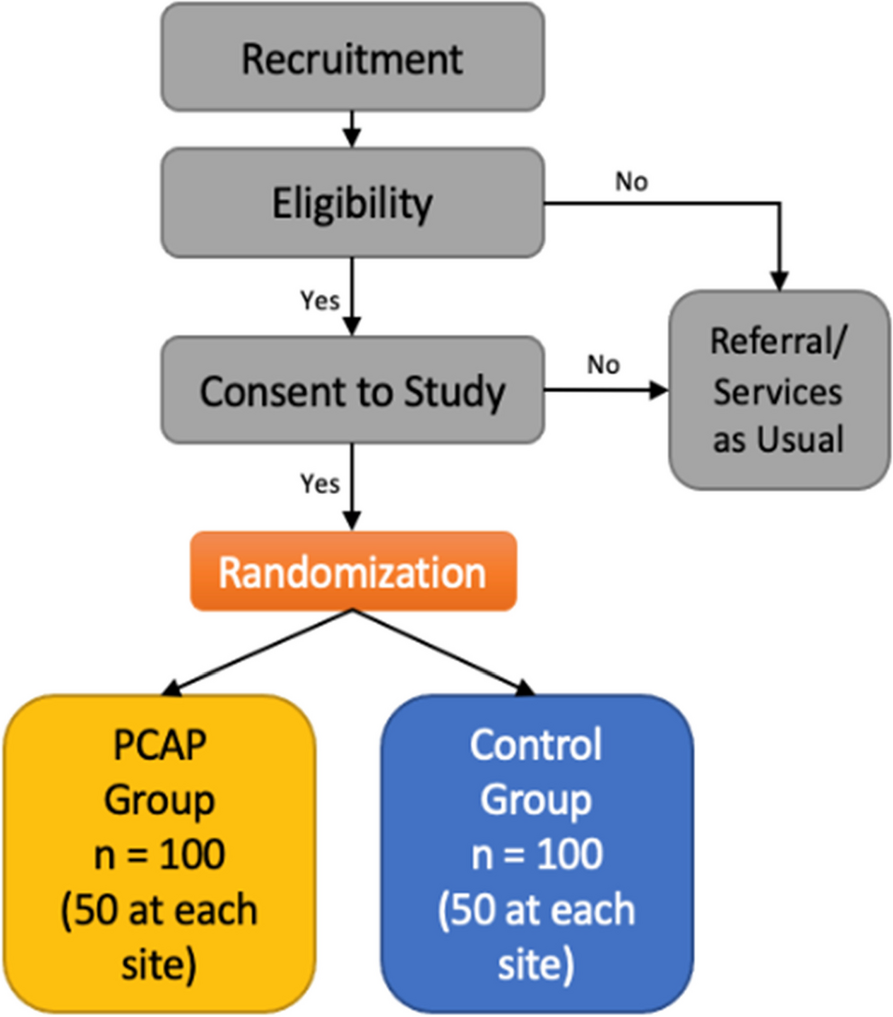
Graphical flowchart of the study design for NS (experiment I and II). In experiment I, patients with Noonan syndrome first receive lamotrigine (LTG) or placebo (PLC) with a minimum pause of 7 days and maximum of 60 days. In experiment II, lovastatin (LOV) or PLC with a minimum pause of 7 days and maximum 60 days. Between experiments, there will be a pause of at least 14 days
Fig. 2
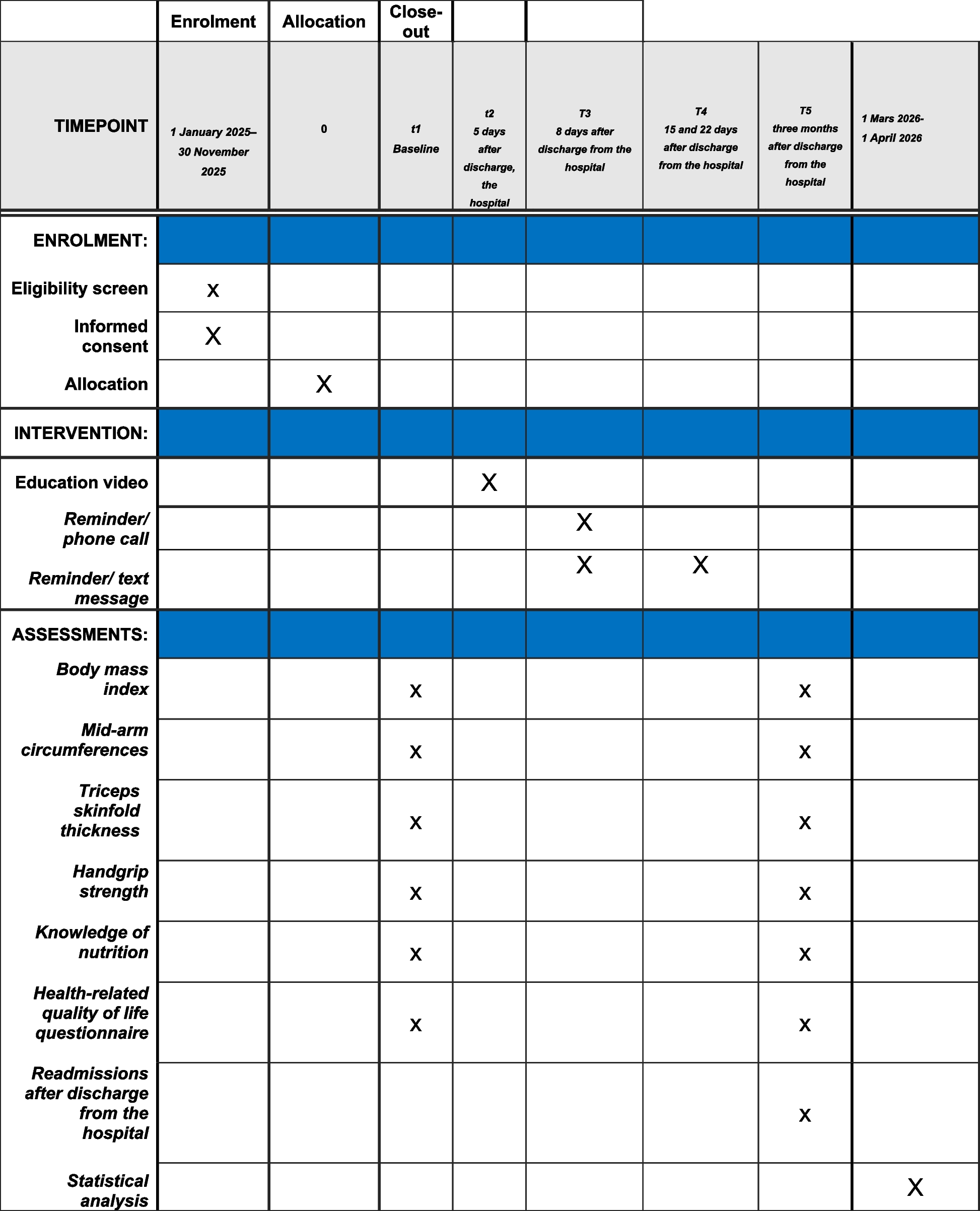
Graphical flow-chart of the study design for NF1 (experiment III). In experiment III, patients with NF1 first receive lamotrigine (LTG) or placebo (PLC) with a minimum pause of 7 days and maximum of 60 days
Trial siteWithin the monocentric setting, the trial site is located at the Department of Social Pediatrics, School of Medicine, Technical University of Munich. Sponsor-Delegated Person is Prof. Dr. med. Volker Mall.
Study populationInclusion criteria are defined as follows: (1) Group 1: NS, Group 2: NF1 (both genetically assured); (2) age ≥ 16 years; (3) signed informed consent of the adolescent (16–17 years of age) who is capable to give his consent and understand the aim and rationale of the study and of the legal guardian; (4) signed informed consent of persons who are ≥ 18 years old and capable to give their consent. In case of doubts, an independent medical practitioner will evaluate the capacity to consent; (5) male and female participants who are not capable of bearing children or who use a method of contraception that is medically approved by the health authority of the respective country.
Exclusion criteria are (1) epilepsy, (2) participants taking medication with known central nervous system (CNS) effects, (3) severe mental retardation, (4) side effects during previous medication with and contraindications for LTG and/or LOV and/or transcranial magnetic stimulation (TMS), (5) psychiatric diseases, (6) previous history of allergic reactions with LTG and LOV medications, (7) potentially unreliable patients, (8) patients who are not suitable for the study in the opinion of the investigator, (9) pregnancy (incl. positive urine pregnancy test) and (10) persons who are incapable of giving consent or do not understand the aim or rationale of the study.
InterventionsParticipants are randomized into either one out of the three approaches (exp. I–III). Three randomizations take place—one for each experiment. Patients are randomly assigned to receive either first verum then placebo, or vice versa. The randomization lists were created using RANCODE professional 2015. Randomization is performed block-wise. The randomization list was provided to the pharmacy for blinding, labelling and distribution of trial medication. For the case of emergency unblinding, a second set of sealed envelopes includes the information on the type of medication for each randomization number and is stored at the trial site. The integrity of the envelopes is monitored until end of study.
Experiment I—Lamotrigine (LTG) in patients with NSParticipants receive 300 mg single dose LTG or placebo 2 h before TMS, corresponding to the peak plasma time of LTG [11]. After at least 7 (up to 60) days, a cross-over takes place: participants receive placebo or 300 mg single dose LTG 2 h before TMS (Fig. 1).
Experiment II—Lovastatin (LOV) in patients with NSParticipants receive 200 mg LOV or placebo daily for 4 days prior to TMS. On the day of TMS, the medication is given 3 h before starting TMS corresponding to the peak plasma concentration of LOV [12]. After at least 14 (up to 60) days, a cross-over takes place: participants receive placebo or 200 mg LOV daily for 4 days prior to TMS (Table 1, Additional file 1). On the day of TMS, the medication is given 3 h before starting TMS.
Table 1 Schedule of procedures and visits patient group 1, NSExperiment III—Lamotrigine (LTG) in patients with NF1Participants receive 300 mg single dose LTG or placebo 2 h before TMS [11]. After at least 7 (up to 60) days, a cross-over takes place: participants receive placebo or 300 mg single dose LTG 2 h before TMS (Table 2, Additional file 1).
Table 2 Schedule of procedures and visits patient group 2, NF1Experiments I and II take place with a sufficient time gap on the same set of patients (Fig. 1). It is not possible to conceal treatment in a triple cross-over design in group 1, which is why two separate tests have been planned on group 1.
Transcranial magnetic stimulation (TMS)Transcranial magnetic stimulation (TMS) is a standard method used in medical diagnostics and research in humans [13, 14]. The planned investigations promise progressing basic findings regarding the neurophysiological mechanisms of the TMS and of neuronal plasticity in patients with NF1 and NS. Planned neurophysiological investigations with TMS delivered by the PowerMAG quadri-pulse stimulation (QPS) device (Mag & More GmbH, Munich) bear only very little risks and have been used by the investigators before [15]. An increased susceptibility to seizures, which can be observed with high-frequency, above-threshold TMS, was not seen so far using the planned stimulation protocols [15]. The clicking noise, which appears using transcranial magnetic stimulation, does not cause any adverse effect. The examination is not painful. Some participants are not used to the involuntary muscle contraction, which appears under magnetic stimulation, and sometimes this is felt as an awkward sensation [14]. However, experience has shown a fast adaption to these contractions. Participants are informed about this beforehand.
No other undesirable side effects are expected during stimulation. The investigators used TMS in patients with NF1 [6] as well as with NS [7] reviewed in [8] to demonstrate deficits in synaptic plasticity which were associated with attention deficits (NF1: [6]). The investigators have a long time experience with TMS and published several manuscripts in international journals [10, 15,16,17]. The study procedure contains pre-measurements before interventional, repetitive TMS using the quadri-pulse theta burst stimulation protocol (qTBS) [15] and three (Post 1–3) follow-up measurements (Fig. 3). Changes in motor-evoked potentials (MEP), referring to the model of long-term potentiation (LTP)-like plasticity, as primary outcome measure, are monitored until 1 h after interventional TMS, conducted by the qTBS protocol. Changes in local excitability are investigated by the resting motor threshold.
Fig. 3
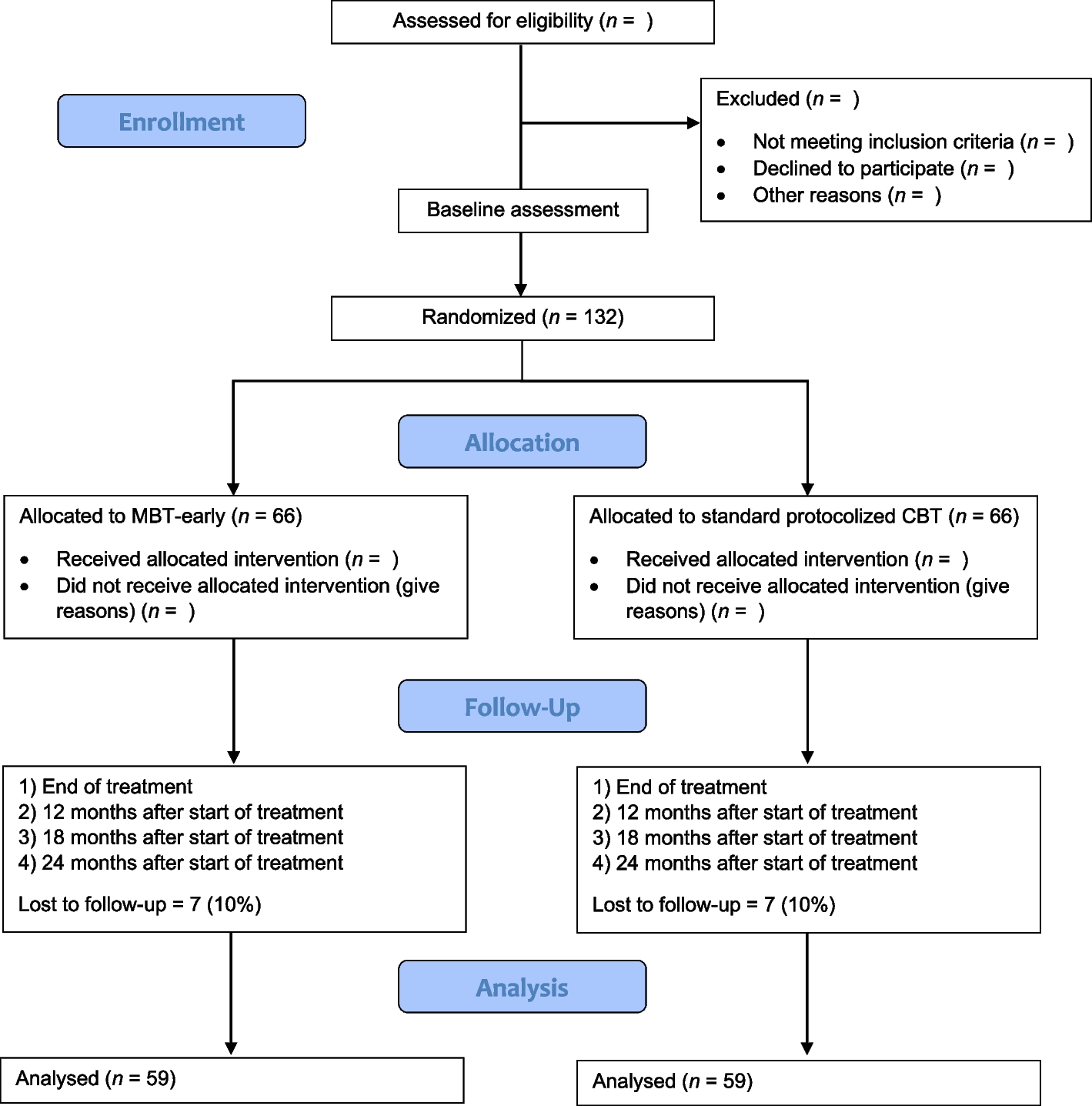
Timeline of TMS measurement. After pre-measurements, patients (NS and NF1) will receive a quadri-pulse theta burst stimulation (qTBS) to evaluate changes in cortico-spinal excitability. Motor evoked potentials (MEP) and resting motor threshold will be monitored 2–5 min, 30 min and 60 min after qTBS. MEP, motor evoked potential; RMT, resting motor threshold; AMT, active motor threshold; qTBS, quadri-pulse theta burst stimulation
Test for Attentional Performance (TAP)The computerized Test for Attentional Performance (TAP) battery is a widely used tool in the systematic assessment of attention [6]. Attention skills of the participants are evaluated by conducting the Alertness, Visual Scanning, GoNogo and Incompatibility task. These four subtests of the TAP were selected because of a strong analogy to attention deficit responsive testing in patients with NF1. The Alertness test is a visual reaction time task to a presented visual target stimulus (cross) with or without previous acoustic warning. The Visual Scanning test requires accuracy and performing speed while detecting the target in a field of similar figures. The GoNogo test assesses the ability to discriminate between a target and a similar but irrelevant non-target, where participants have to respond to the target and inhibit their response to the non-target. In the Incompatibility test, participants have to detect the direction of a simple arrow (right/left) by pressing a button on the left or right side regardless on which side of the screen (right/left) the arrow appears.
Endpoints and endpoint rationaleThe primary endpoint for each experiment is the difference between the amplitude of the motor evoked potential (MEP) elicited with transcranial magnetic stimulation (TMS, measured at three time points after interventional TMS for each investigation) after placebo and after medication (LTG and LOV).
Changes in MEP are a well-established and safe method to probe changes in cortico-spinal excitability in humans [18]. MEPs are elicited by TMS using a figure-of-eight coil with an outer diameter of 100 mm centred tangentially on the scalp over the primary motor cortex (M1) of the nondominant hand with its handle pointing in a posterior direction and laterally at an angle of approximately 45° away from the midline. MEPs are recorded from the abductor pollicis brevis (APB) muscle at rest by surface electromyography (EMG) using silver/silver chloride electrodes with a surface area of 263 mm2 (AMBU, Ballerup, Denmark) mounted in belly-tendon recording technique. Data are band-pass filtered (20–2000 Hz) and amplified using an Ekida DC universal amplifier (EKIDA GmbH, Helmstadt, Germany), digitized at 5 kHz sampling rate using a MICRO1401mkII data acquisition unit (Cambridge Electronic Design Ltd, Cambridge, UK) and stored on a standard personal computer for online visual display and later offline analysis using Signal Software version 5 (CED Ltd, UK). MEP size is determined by measuring the two highest peaks of opposite polarity. Twenty trials will be recorded and then averaged for each point of investigation as described elsewhere [15, 16]. Resting motor threshold is recorded to probe changes in local cortical excitability [19].
The secondary endpoints for each experiment include the difference between the neuropsychological testing of attention by the TAP and differences in short interval cortical inhibition (SICI) after placebo and after medication (LTG and LOV) [6, 20]. Another endpoint is the comparison of LTG and LOV effects on synaptic plasticity and attentional performance in the NS group. The TAP is a well-established tool and used to measure the influence of LTG and LOV on attention [7, 20]. We previously demonstrated that LOV leads to an amelioration of attentional performance in humans with NF1 [6]. Consequently, we hypothesized that this would also lead to better attention in patients with NS and that LTG ameliorates attention in NF1 and NS.
For SICI, a subthreshold conditioning stimulation is delivered 2, 3 and 5 ms before a test stimulus [6]. It has been demonstrated before that an increased inhibition is the most prominent factor in mice with NF1 to prevent synaptic plasticity [5]. The authors have previously demonstrated that LTG is a potent modulator of synaptic plasticity in healthy humans [10]. Decreasing intracortical inhibition by LOV was associated with an increase in synaptic plasticity in patients with NF1 [6]. The central role of intracortical inhibition regulating synaptic plasticity in NF1 has recently been certified by animal studies revealing a new option of pharmacological intervention with LTG [5]. We have previously shown that LOV leads to a disinhibition in patients with NF1 and, therefore, hypothesized that this would be a key factor to normalize cortico-spinal excitability in patients with NF1 and NS.
Safety measures include documentation of blood pressure and heart frequency prior to the intervention as well as monitoring of EMG activity during TMS intervention and documentation of adverse events (AE), severe adverse events (SAE) and suspected unexpected serious adverse reactions (SUSAR) following established definitions and legal requirements. The intensity of AEs is defined according to the common terminology criteria for adverse events (CTCAE Version 4.0, https://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf).
Analyses and statisticsThis is a series of three experiments in a double-blind, placebo-controlled, randomized, cross-over design. Exp. I: LTG vs. placebo on group 1, Exp. II: LOV vs. placebo on group 1, Exp. III: LTG vs. placebo on group 2.
Sample size: Sample size calculation was done using the primary endpoints, nQuery Advisor, and expected means of MEP amplitudes and standard deviation (SD) for the different measurements from Mainberger et al. [7].
Timepoint 1 (T1)
Timepoint 2 (T2)
Timepoint 3 (T3)
Lovastatin LTP (increase in mean MEP) data (Mainberger et al. [7]) with common SD of 0.5
Healthy controls
1.440
1.600
1.710
NF1
0.920
0.970
0.980
Difference
0.520
0.630
0.730
Experiments I and II
Local α
0.025
0.013
0.008
Power with 14 patients
89%
95%
97%
Power with 12 patients
82%
89%
94%
Experiment III
Local α
0.050
0.025
0.017
Power with 14 patients
94%
97%
99%
Power with 12 patients
90%
94%
97%
An increase in MEP amplitudes over time is considered to refer to the concept of long-term potentiation (LTP)-like plasticity as it has been demonstrated in Mainberger et al. [7] as well. Twelve patients per disease entity will be enough to provide appropriate power to all planned primary endpoint tests (two-sided paired samples t-tests) with a global significance level of 5% and adjusted local significance levels using the Bonferroni-Holm procedure. In order to account for some unobtainable data and drop outs, the sample size will be increased to 14 patients per disease (28 trial participants in total).
Analysis sets: For each experiment, there will be a separate analysis set defined, as it is possible that a patient is suitable for analysis in only one of the first two experiments. All analyses will be performed on the full analysis set (FAS-I, FAS-II, FAS-III), consisting of all patients who delivered a full set of MEP measurements within the corresponding experiment. Since all measurements will take place within a very short time per patient, it is expected that the data will be either complete or entirely missing.
Primary endpoint: The primary endpoint analyses will be performed in three separate testing procedures. The global significance level will be 5%. Since experiments I and II are done on the same set of patients, the significance level for those two experiments will each be 2.5%. The significance level of experiment III will be 5%. The primary endpoint analysis will consist of three series of three paired samples two-sided t-tests, comparing MEP under verum vs. placebo at the three measurement time points. The local significance level will be adjusted using the Bonferroni-Holm procedure and will be as follows for the ordered by p-value tests:
Experiment
Test number
Local significance level
I and II
1
0.025
I and II
2
0.013
I and II
3
0.008
III
1
0.050
III
2
0.025
III
3
0.017
Secondary endpoints and safety: Analyses of baseline and safety data and secondary endpoints will be done using appropriate descriptive statistics and paired sample tests for difference between the two study groups. All tests will be two-sided with an exploratory significance level of 5%. No adjustment for multiple comparisons will be done.
Organizational frameworkOrganizational/regulatory project management, safety management, monitoring and data management are performed by the Münchner Studienzentrum (MSZ), Technical University of Munich, School of Medicine. An independent safety monitoring board (SMB) is established. The underlying principles for the SMB are ethical and safety aspects for the patients. The SMB examines whether the conducting of the study is still ethically justifiable, whether safety of the patients is ensured and whether the process of the study is acceptable. For this, the SMB is informed regularly about patient recruitment and observed safety advents. Serious adverse events (SAEs) are recorded in a study-specific safety form and transferred into a safety database by the safety management, which processes further SAE documentation in written form to the SMB, the Investigator, the ethics committee and regulatory authority (BfArM) according to applicable law.
All study procedures agree with the guidelines of Good Clinical Practice (GCP) of the International Council on Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) and the principles of the Declaration of Helsinki. All participating investigators agreed to adhere to the instructions and procedures described in the study protocol and thereby to adhere to the principles of ICH-GCP. All members of the safety board are independent from the sponsor. The current approved protocol version is 5.0 (amendment 3, 21.09.2020). All protocol versions are available in German language at nikolai.jung@tum.de.
留言 (0)