
記住我
Wolf–Hirschhorn syndrome (WHS; OMIM 194190) is a rare genetic syndrome caused by terminal chromosome 4p deletions and is also known as 4p-syndrome. WHS is characterized by typical craniofacial features including a “Greek warrior helmet” appearance of the nose (wide flattened nasal bridge continuing to the forehead), microcephaly, widely spaced eyes, a distinct mouth, a short philtrum, micrognathia, downturned corners of the mouth, and poorly formed ears with pits and tags. Growth restriction, postnatal growth deficiency, hypotonia with muscle underdevelopment, and developmental delay/intellectual disability of variable degrees are observed in all affected patients. Other findings include heart and skeletal defects, seizures, abnormal tooth development, and hearing loss (1,2).
WHS is caused by a deletion in the 4p16.3 region, with deletions shorter than 3.5 Mb associated with a milder phenotype without major malformations, and two WHS critical regions—WHSCR−1 and WHSCR−2—overlap the WHSC1 gene (3–5). The WHS candidate 1 (WHSC1) gene, also known as NSD2 (OMIM 602952), encodes nuclear receptor-binding set domain protein 2 and play a important role in normal development (6). Recently, 36 novel or de novo variants in NSD2 variants were identified in 36 patients with overlapping yet atypical features of WHS, thus defining a novel WHS-like disorder, namely Rauch-Steindl syndrome (RSS,OMIM 619695) (Supplementary Table S1) (7–12). Patients with RSS exhibit a wide range of mild phenotypic features, with core manifestations of microcephaly, intrauterine growth restriction, facial dysmorphisms, autism, intellectual disability, and low birth weight, feeding difficulties, failure to thrive, short stature, speech delay and muscular hypotonia.In this study, In this study, we identified a novel de novo NSD2 gene variant [c.2721delT(p.Asn907Lysfs*5)] in a Chinese girl diagnosed with Rauch–Steindl syndrome. In addition, our study further expands the phenotypic spectrum of WHS and the mutation spectrum of NSD2.
Materials and methods PatientsA 7 year-old-girl was referred to the Genetic Department of Guangxi Maternal and Child Health Hospital for delayed development and growth on November 17th, 2019 (Figure 1). The proband's parents provided written informed consent for the publication of photographs as well as clinical and genetic data. The present study was approved by the Department of Genetic Metabolic Central Laboratory of Guangxi Zhuang Autonomous Region, Women and Children Care Hospital.
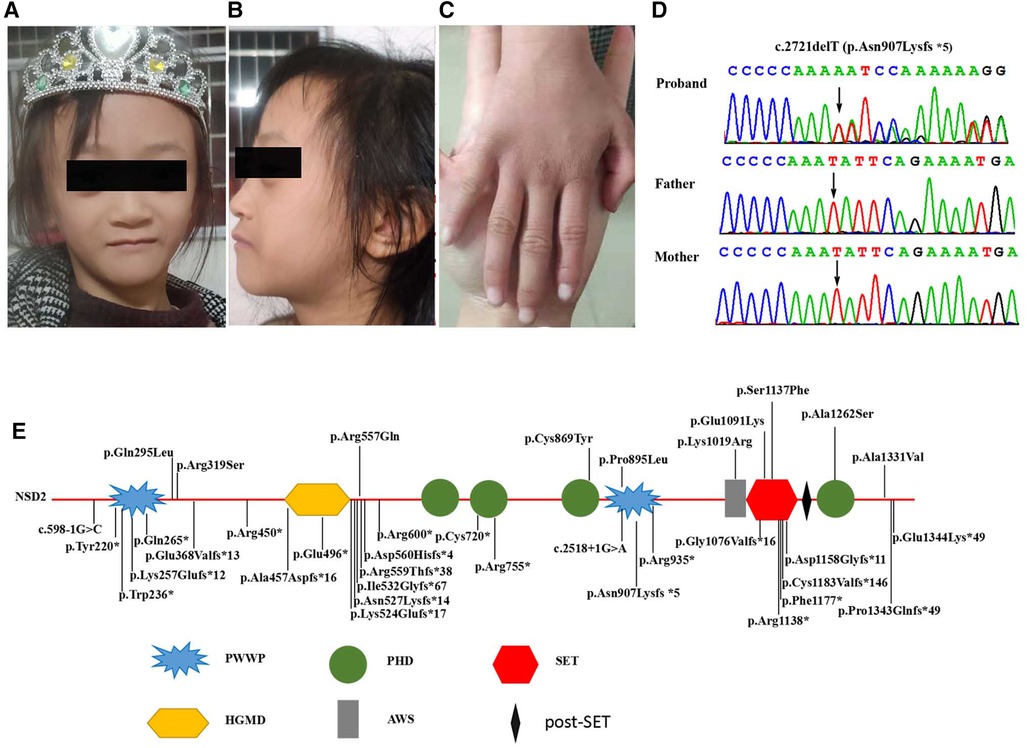
Figure 1. (A,B) Clinical photographs of proband showing triangular face, proptosis, hypertelorism, prominent nasal bridge continuing to forehead, micrognathia. (C) Mild clinodactyly was observed on her right hand. (D) Sequence chromatograms of the de novo truncating variant in NSD2 with their locations indicated by black arrow. (E) Schematic diagram of the location of the NSD2 gene domains and variants. (PWWP, Pro-Trp-Trp-Pro conserved motif; HMG, High mobility group box; PHD, Plant Homeodomain finger; AWS, associated with SET domain; SET, suppressor of variegation, enhancer of zeste, and Trithorax domain).
Genetic analysis Whole-exome sequencing and Sanger sequencingGenomic DNA was extracted from 2-ml peripheral blood samples from the proband. Whole exome sequencing (WES) was performed at the department of Genetic and Metabolic Central Laboratory, Guangxi Maternal and Child Health Hospital. Target DNA was captured using the Agilent SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's instructions, and the captured libraries were sequenced with the Illumina HiSeq 2500 system (Illumina). The Translational Genomics Expert (TGex) platform (LifeMap Sciences, USA) with the VarElect scoring system was used to annotate the selected variants (13).
In silico analysisSIFT, PolyPhen 2.0, and Mutation Taster software were used to calculate the pathogenicity index of all novel missense variants with unknown clinical significance. The candidate NSD2 variant identified by WES was confirmed by Sanger sequencing, and its pathogenicity was classified following to American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (14).
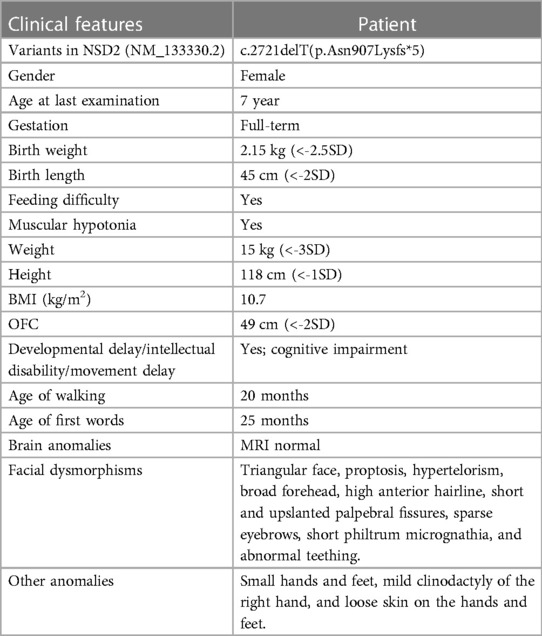
Results Clinical descriptionThe proband is a 7 seven-year-old girl born to healthy parents as the first child. Her non-consanguineous parents originated from Guangxi, China. She was admitted to the department of Pediatric Genetic and Metabolic Central Laboratory of Guangxi Maternal and Child Health Hospital due to delayed development and growth. She was born at full term with a birth weight of 2.15 kg and suffered from intrauterine growth restriction during pregnancy. Low birth weight and feeding difficulties kept her hospitalized for a long time. She began walking around 20 months, and spoke her first sentences around 25 months. Growth restriction persisted, she had severe malnutrition [weight: 15 kg (<-3 SD), BMI: 10.7 kg/m2], and presented with mild short stature (height: 118 cm, <-1 SD). Her facial dysmorphic features included microcephaly (head circumference: 49 cm, <-2 SD), a triangular face, proptosis, hypertelorism, a broad forehead, a high anterior hairline, short and upslanted palpebral fissures, sparse eyebrows, short philtrum micrognathia, and abnormal teething (Table 1). Dysmorphic features also included small hands and feet, mild clinodactyly of the right hand, and loose skin on the hands and feet. Evaluation using the Chinese Webster Intelligence Scale showed that her full IQ was 85 (Table 1). She had no behavioral issues. The patient's brain MRI, EEG, x-ray of the chest and spine, echocardiography, and abdominal ultrasound examinations were normal, and she experienced no seizures. Her karyotype was determined to be 46, XX.
Table 1. Clinical features of the patient with de novo NSD2 variant.
Mutation analysisWe performed WES for the proband at an average depth of coverage of 20×, and 95.6% of the targeted regions were covered. A total of 132,342 variants or indels were detected in the proband. Variants with a minor allele frequency of >0.01 in any of the variant databases (e.g., 1,000 Genomes, dbSNPand Genome Aggregation Database)were excluded.Using TGex software (LifeMap Sciences, United States), ten candidate variants matched with known phenotypes in ten genes (FGFR3, PCNT, CDC45¸UBE3B, TBCE, NSD2, CPLX1, SETD5, NOTCH2, FZD2) were subsequently extracted. The variants in the PCNT, CDC45, UBE3B, TBCE and CPLX genes were heterozygous. The diseases caused by these genes are autosomal recessive and are therefore excluded. The variants of the FGFR3, SETD, NOTCH2 and FZD2 genes were inherited from the unaffected father or mother, but were found not to be responsible for the phenotype (Supplementary Table S2). Then, a novel de novo heterozygous frameshift variant c.2721delT(p.Asn907Lysfs*5) in the NSD2 gene (RefSeq: NM_133330.2) was identified in the proband (Figure 1D). The variant has not been previously reported in 1,000 Genomes, dbSNP, Genome Aggregation Database and in control databases. According to the ACMG/AMP standards and guidelines for the variant, the novel variant is pathogenic (PVS1, PS2, PM2).
Discussion and conclusionNSD2 (WHSC1) is∼90 kb long and located on human chromosome 4p16.3, and encods the nuclear SET domain-containing transcriptional regulatory protein, which contains four development-related domains: a PWWP domain, an HMG box, a SET domain, and a PHD-type zinc finger. NSD2 is a SET domain histone methyltransferase responsible for the methylation of H3K36, is expressed widely across many tissue types, and participates in a variety of biological processes, including early development, cytokine signaling, the DNA damage response, and class switch recombination. Haploinsufficiency of NSD2 (pLi = 1.00) is thought to be to be an important part of the mutational mechanism of WHS. Heterozygous knockout-NSD2 mouse models show growth restriction, craniofacial malformation, and midline fusion defects (15, 16). Zanoni et al. demonstrated that loss-of-function and missense variants in NSD2 lead to reduced methylation activity and are associated with a distinct developmental phenotype (11). These findings suggest that NSD2-deficiency is responsible for multisystem abnormalities in patients with Rauch–Steindl syndrome. Recent reports have confirmed that patients with novel loss-of-function or missense variants in NSD2 exhibit a variety of abnormalities including facial dysmorphism, microcephaly, intrauterine and postnatal growth restriction, craniofacial malformations, failure to thrive, intellectual disability, speech delay, behavioral/psychological issues, and skeletal and limb abnormalities (7–12). In the present study, the de novo variant c.2721delT is located in the PWWP2 domain in NSD2 and is predicted to create a frameshift starting at codon 907 leading to a stop codon 5 positions downstream (p.Asn907Lysfs*5). This variant may result in the absence of protein production with a significant decrease in mRNA level due to nonsense-mediated decay degradation. The present patient showed common phenotypes associated with Rauch–Steindl syndrome, including facial dysmorphism, microcephaly, a degree of intellectual disability, growth restriction, severe malnutrition, short stature, small hands and feet, muscular hypotonia, mild clinodactyly of right hand, and loose skin on the hands and feet, and these phenotypes are thought to be due to the loss-of-loss-of-function variant detected in NSD2.
To date, 37 NSD2 variants have been identified, including ten missense variants (eight de novo, two inherited), fifteen frameshift variants (nine de novo, two inherited, four undetermined), ten nonsense variants (nine de novo, two undetermined), and two splice site variants (two de novo) (Supplementary Table S1). These variants are distributed across the entire gene, and 87.5% (28/32) are de novo (Supplementary Table S1, Figure 1). Of the 37 NSD2 variants, 11 (including our variant) located on the PWWP domain, including two missense variants (two de novo), four frameshift variants (three de novo, one undetermined), and four nonsense variants (three de novo, one undetermined). This is not significant compared to the variants in the entire gene. In addition, Rauch-Steindl syndrome is a highly heterogeneous disease caused by variants in the NSD2 gene. The phenotypes of the Rauch-Steindl syndrome are complex but several common characteristics are observed in most patients (11). Zanoni et al. have been reported that the core NSD2-associated phenotype includes mostly mild developmental delay, prenatal-onset growth retardation, low body mass index, and characteristic facial features distinct from WHS (11). Patients carrying missense variants were significantly taller and had more frequent behavioral/psychological issues compared with those harboring truncating variants. The results showed that the relationship between genotype and phenotype seemed to be related to the mutation type of the mutant, but not to the position of the mutant in the protein. Limited by the currently reported cases and variants, these results should be seen as provisional. Future studies with the report of increasing numbers of patients will therefore be necessary to to refine the phenotype and clarify genotypic effects and other phenotypic determinants.
Typical WHS has been described as a disorder with special craniofacial features, growth restriction, intellectual disability, seizures and skeletal abnormalities resulting from heterozygous deletions in the 4p16.3 region (1). Compared with typical WHS patients, an atypical mild manifestation was observed in our patient. In addition to our patient, other reported patients showed less marked craniofacial features of WHS. 60%–70% of typical WHS patients have severe skeletal anomalies including kyphosis/scoliosis with malformed vertebral bodies, accessory or fused ribs, clubfeet, and split hands. Only 39% of patients with a NSD2 variant have skeletal abnormalities, which is less severe than in typical WHS patients (9–11). Mild clinodactyly in the right hand of the present patient was observed. This indicates that the NSD2 gene may not be the main cause of WHS craniofacial features and skeletal anomalies, and it may have a cumulative effect on severe intellectual disability, typical craniofacial features, and skeletal anomalies of WHS with other genes located in the 4p16.3 region. Seizures are the most common problem in children with WHS (90%–100%) and are the greatest problems in the clinical management of WHS (17). As in previously reported patients with NSD2 variants, our patient also lacked seizures or seizure-like episodes, further indicating that seizures are not associated with the NSD2 gene and that other genes located within the 4p16.3 region such as LETM1 may be responsible for seizures (18). In addition, the severity of the intellectual disability in patients with NSD2 patients was milder compared with patients carrying the most common 4p deletions (which are between 5 and 18 Mb in length). Furthermore, the majority of individuals with NSD2 variants lack many of the most common manifestations of WHS, such as orofacial clefts and defects and genital, cardiac, and renal malformations. Therefore, NSD2-deficiency may be necessary for WHS, but it is not sufficient to cause WHS. In addition, genetic diagnosis is the best method for detecting the cause of the syndrome in these patients.
ConclusionsIn conclusion, we found a de novo heterozygous frameshift pathogenic variant, c.2721delT(p.Asn907Lysfs*5), in the NSD2 gene in a Chinese girl with Rauch–Steindl syndrome using WES. Our findings support that this loss-of-function variant in NSD2 is a cause of a syndrome consisting of intellectual disability and developmental delay. Detailed clinical features and molecular diagnosis will further help our understanding of the phenotype-genotype correlations of NSD2 pathogenic variants and related disorders including WHS. In addition, molecular genetic testing of NSD2 is a useful tool for clinical diagnosis and genetic counseling of these patients. The novel pathogenic variant uncovered in this study expands the mutation spectrum of NSD2.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/, PRJNA888339.
Ethics statementThe studies involving human participants were reviewed and approved by Institutional Review Boards and Ethics Committees of Guangxi Maternal and Child Health Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributionsQY and QZ designed the present study. DG, SY and JL were responsible for clinical information collection, experiments and data analysis. QY and QZ were responsible for manuscript writing and revision. All authors contributed to the article and approved the submitted version.
FundingThis work was supported by the Health Department of Guangxi Province (Grant Nos. Z-A20220256) and Guangxi Key Research and Development Program (GuikeAB17195004), which have been used for the patient recruitment and deter-mining genetic variants. Guangxi Key Laboratory of Birth Defects and Stem Cell Biobank (ZTJ2020002).
AcknowledgmentsWe are grateful to the family for participating in this study.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material.The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1064783/full#supplementary-material.
AbbreviationsWHS, Wolf–Hirschhorn syndrome; WHSC, Wolf–Hirschhorn syndrome critical region; WHSC1, Wolf–Hirschhorn syndrome candidate 1; WES, Whole exome sequencing; TGex, Translational Genomics Expert; ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology.
References1. Battaglia A, Carey JC, South ST. Wolf-Hirschhorn Syndrome—Retired Chapter, for Historical Reference Only. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. Genereviews®. Seattle, WA: University of Washington . (2015).
3. Wright TJ, Ricke DO, Denison K, Abmayr S, Cotter PD, Hirschhorn K, et al. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum Mol Genet. (1997) 6:317–24. doi: 10.1093/hmg/6.2.317
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. (2003) 72(3):590–7. doi: 10.1086/367925
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Kim JY, Kee HJ, Choe NW, Kim SM, Eom GH, Baek HJ, et al. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol Cell Biol. (2008) 28(6):2023–34. doi: 10.1128/MCB.02130-07
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Barrie ES, Alfaro MP, Pfau RB, Goff MJ, McBride KL, Manickam K, et al. De novo loss-of-function variants in NSD2 (WHSC1) associate with a subset of Wolf-Hirschhorn syndrome. Cold Spring Harb Mol Case Stud. (2019) 5:a004044. doi: 10.1101/mcs.a004044
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Lozier ER, Konovalov FA, Kanivets IV, Pyankov DV, Koshkin PA, Baleva LS, et al. De novo nonsense mutation in WHSC1 (NSD2) in patient with intellectual disability and dysmorphic features. J Hum Genet. (2018) 63:919–22. doi: 10.1038/s10038-018-0464-5
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Jiang Y, Sun H, Lin Q, Wang Z, Wang G, Wang J, et al. De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype. BMC Med Genet. (2019) 20134. doi: 10.1186/s12881-019-0863-2
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Derar N, Al-Hassnan ZN, Al-Owain M, Monies D, Abouelhoda M, Meyer BF, et al. De novo truncating variants in WHSC1 recapitulate the Wolf-Hirschhorn (4p16.3 microdeletion) syndrome phenotype. Genet Med. (2019) 21:185–8. doi: 10.1038/s41436-018-0014-8
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Zanoni P, Steindl K, Sengupta D, Joset P, Bahr A, Sticht H, et al. Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet Med. (2021) 23:1474–83. doi: 10.1038/s41436-021-01158-1
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Hu X, Wu D, Li Y, Wei L, Li X, Qin M, et al. The first familial NSD2 cases with a novel variant in a Chinese father and daughter with atypical WHS facial features and a 7.5-year follow-up of growth hormone therapy. BMC Med Genomics. (2020) 13:181. doi: 10.1186/s12920-020-00831-9
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, et al. Varelect: the phenotype-based variation prioritizer of the GeneCards suite. BMC Genomics. (2016) 17444. doi: 10.1186/s12864-016-2722-2
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Estabrooks LL, Rao KW, Driscoll DA, Crandall BF, Dean JC, Ikonen E, et al. Preliminary phenotypic map of chromosome 4p16 based on 4p deletions. Am J Med Genet. (1995) 57:581–6. doi: 10.1002/ajmg.1320570413
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. (2009) 460:287–91. doi: 10.1038/nature08086
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Battaglia A, Filippi T, Carey JC. Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet. (2008) 148C:246–51. doi: 10.1002/ajmg.c.30187
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Monies D, Abouelhoda M, AlSayed M, Alhassnan Z, Alotaibi M, Kayyali H, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. (2017) 136:921–39. doi: 10.1007/s00439-017-1821-8
留言 (0)