
記住我
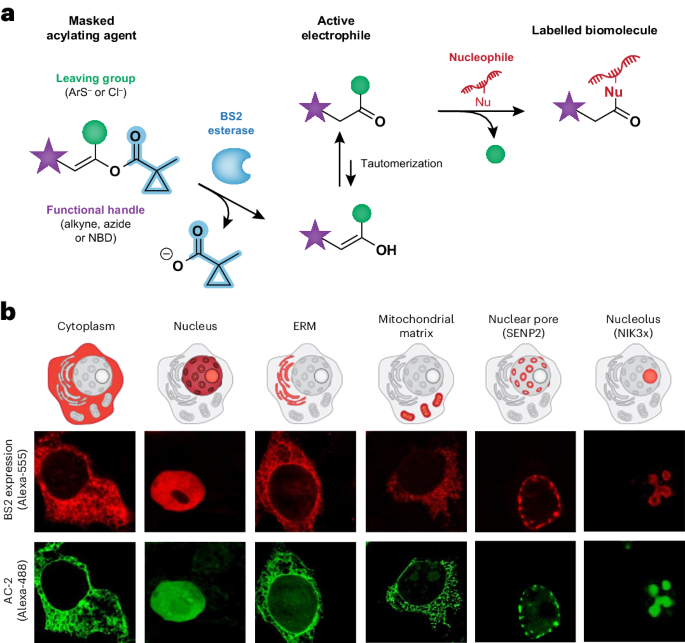
As a result of the high nucleophilicity of the 6p2 lone pair on the Bi(I) centre, bismuthinidene 1 (refs. 37,38) has recently been shown to engage in polar SN2-type reactions with alkyl halides and triflates39. Similarly, 1 reacted quantitatively with a range of benzyl (pseudo)halides (Cl, Br, I, mesylate) to give benzyl bismuth(III) complexes 5–8 (Fig. 2b). Cyclic-voltammetry analysis of 1 (E1/2 = −0.85 versus Fc0/+, the ferrocene/ferrocenium couple) provides evidence that C‒X (X = halide) cleavage should proceed through a classical SN2 pathway (Ep/2 < −2.0 V versus Fc0/+). On the other hand, the electrochemical behaviour suggested that 1 could potentially engage in SET oxidative-addition processes with alkyl redox-active electrophiles (Fig. 2a). Accordingly, reaction of 1 with 1 equiv. of tetrachlorophthalimide (TCPhth) ester 2 (Ep/2 = −1.2 V versus Fc0/+) cleanly afforded benzyl bismuth(III) complex 9 after SET, fragmentation, release of CO2 and radical recombination (given that the potential difference between 1 and 2 is approximately 0.35 V, SET between 1 and 2 can be estimated to be approimately 8 kcal mol−1 uphill, but subsequent release of CO2 can drive the oxidative-addition process)40. The resulting alkyl-bismuth(III) adduct could be fully characterized by NMR, high-resolution mass spectrometry (HRMS) and single-crystal X-ray diffraction. Furthermore, KS 4 (Ep/2 = −1.3 V versus Fc0/+) also underwent radical oxidative addition with 1 to give 10. As expected, non-chlorinated phthalimide ester 3 (Ep/2 = −2.0 V versus Fc0/+) remained unreacted when mixed with 1. Besides benzyl groups, the same process occurs with primary (12) or secondary (13) RAEs, leading to stable alkyl-bismuth(III) complexes. Tertiary RAEs such as the one derived from 1-adamantanecarboxylic acid did also react with 1, but the resulting adducts were found to be unstable and could not be characterized under standard conditions. Interestingly, the process is orthogonal to classical polar transition-metal oxidative additions, as it could be performed in the presence of an aryl bromide, giving 11 as the sole product in 93% yield (Fig. 2b). This reactivity is a rare example where bismuth, besides emulating the redox behaviour of first-row transition metals during oxidative addition, allows the isolation and characterization of the corresponding alkyl‒metal species resulting from radical recombination. We also found that, whereas classical SN2 reactivity is sensitive to steric effects (>24 h for 14), single-electron oxidative addition of the corresponding RAE led to quantitative formation of complex 12 in <5 min. Furthermore, we found complexes 9, 12 and 13 to be active by electron paramagnetic resonance (EPR) spectroscopy, especially upon light irradiation. Low-temperature EPR analysis of 12 suggests the formation of two radical species that decay at different rates. This is consistent with the homolysis of the C–Bi bond (see Supplementary Information for details)41,42. To investigate this behaviour further, the reaction of bismuthinidene 1 with cyclopropylmethyl iodide was monitored by NMR at low temperature in the dark (Fig. 2c). Complete conversion into cyclopropylmethyl adduct 16 was observed within 1 h at −20 °C. When the mixture was warmed to 50 °C, a slow but steady conversion to ring-opening compound 18 was observed (35% after 12 h).
Fig. 2: Oxidative additions to bismuth(I).
a, Evaluating electronically different (polar, Ep/2 < –2.0 V, versus radical, Ep/2 > –2.0 V) oxidative additions to bismuthinidene 1 (left). Cyclic voltammetry of 1 (right). b, Stable oxidative-addition complexes accessed via SN2 (5–8 and 14) or SET (9–13) mechanisms. c, Evidence for alkyl-radical formation after oxidative addition. a Cyclic voltammetry recorded in MeCN, potential in V versus Fc0/+. b Cyclic voltammetry recorded in MeCN (ref. 47); potential in V versus Fc0/+ converted from V versus saturated calomel electrode (−2.13 V). c Yields and conversions determined by 1H NMR, unless noted otherwise. Ts, 4-toluenesulfonyl; MsO, mesylate.
This indicates that homolysis of the Bi‒C bond in 16 takes place, leading to an in-cage radical pair (int-1). Furthermore, subjecting a solution of 16 to blue light-emitting diode (LED) irradiation resulted in complete conversion into open product 18 within 5 min, showing that light can accelerate the radical ring-opening process25. Conversely, when cyclopropylmethyl RAE 17 was reacted with bismuthinidene 1, a complex analogous to 16 was not observed; instead, radical ring-opening product 19 was immediately obtained, even in the dark. This is consistent with the two distinct mechanistic scenarios for the oxidative addition. On one hand, polar SN2-type reaction of cyclopropylmethyl iodide with 1 initially leads to 16, which eventually ring-opens via alkyl-radical formation. On the other hand, SET and fragmentation of RAE 17 lead to an in-cage bismuth(II)/alkyl radical pair (int-1), for which cyclopropane ring-opening is faster than radical recombination, resulting in the formation of 19 (Fig. 2b). This alkyl radical-type reactivity is consistent with the behaviour displayed by these complexes: the secondary alkyl radical derived from 13 reacts with Michael acceptors such as phenylvinylsulfone giving Giese addition product 21, either in the dark (57%) or under blue-light irradiation (85%) (Fig. 3a, left). Additionally, the alkyl fragment of several complexes reacted with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO) leading to C(sp3)‒TEMPO adducts. Moreover, we observed that catalytic amounts of 1 can promote Giese-type reactions, among others, upon blue-light irradiation (Supplementary Information)43. When investigating the stability of the Bi(III)‒alkyl compounds in solution, it was found that benzyl bismuth(III) complex 9 was especially sensitive to light irradiation, resulting in decomposition mainly to benzyl–benzyl dimers and unselective benzylation of the N,C,N ligand. On the other hand, complex 13 was stable in solution, even after 3 days at 60 °C (Fig. 2b)44. However, under blue-LED irradiation, 13 underwent slow but clean conversion into elimination product 15 and Bi(I), in a radical-type elimination reminiscent of that of alkylcobaloximes45. Furthermore, scrambling experiments confirm that the exchange of alkyl fragments between two different Bi(III) adducts is also possible (Supplementary Information). Interestingly, when attempting the isolation of α-amino alkyl-bismuth(III) adduct 23 derived from proline, we observed the exclusive formation of the product of decarboxylative amination 24, with recovery of bismuthinidene 1 (Fig. 3a, right)30,33. It was speculated that product 24 would arise from the oxidation of the corresponding α-amino alkyl radical by a highly reactive bismuth(II) species. This would lead to the formation of an electrophilic iminium ion28, which ultimately reacts with the TCPhth anion to make the C‒N bond (Fig. 4 and Supplementary Information).
Fig. 3: Divergent reactivity of an unbiased alkyl-bismuth complex and an α-amino alkyl-bismuth complex.
a, Stable unbiased alkyl-bismuth(III) intermediates displaying typical alkyl-radical reactivity (left) and unstable α-amino alkyl-bismuth(III) complexes that evolve into iminium ion intermediates upon release of bismuth(I) (right). b, Development of a bismuth-catalysed C–N cross-coupling reaction based on the oxidation of α-amino alkyl radicals. aStandard reaction conditions: 22 (1 equiv.) and 25 (3 equiv.) in the presence of bismuthinidene 1 (10 mol%) in DMA (0.033 M) at 25 °C for 2 h. Yields determined by 1H NMR using diphenylmethane as internal standard. Ts, 4-toluenesulfonyl; Boc, tert-butoxycarbonyl.
Fig. 4: Proposed mechanistic rationale.
The C–N cross-coupling reaction of α-amino and α-oxo acids via bismuth(I)-catalysed SET. The graph shows EPR analysis of 23. R3R4NH, NH-heterocycle; OA, oxidative addition; Boc, tert-butoxycarbonyl.
At this point, it was envisaged that this reactivity could exploit the resulting iminium intermediates with external N-nucleophiles, leading to a formal C–N cross-coupling reaction. After optimization of the conditions, we found that the reaction of RAE 22 with 3 equiv. of benzimidazole (25) in the presence of 10 mol% of 1 in dimethylacetamide (DMA) at 25 °C afforded the product of C–N cross coupling (26) in 88% yield within 2 h (Fig. 3b). Under these conditions, the only observed side products were 24 (nucleophilic competition by TCPhth) and the expected amide bond-formation product 27, which could be minimized by controlling the stoichiometry and selecting the appropriate solvent (Fig. 3b, entry 1 versus entry 3) (see Supplementary Information for optimization details). Control experiments without the Bi catalyst led exclusively to an acyl-transfer product (Fig, 3b, entry 2). The high efficiency of the optimized reaction relies on the faster kinetics of the Bi-catalysed radical reaction compared to the background amide formation. For example, the reaction could be carried out at −30 °C in DMF (Fig. 3b, entry 4) or at room temperature in DMA in only 10 min (Fig. 3b, entry 5), giving the desired product in 56 and 85% yield, respectively. To exclude completely the requirement of photoexcitation for any of the steps of the transformation to proceed, the reaction was carried out under exclusion of ambient light, giving comparable results (Fig. 3b, entry 6). As expected, the addition of TEMPO completely inhibited the reaction (Fig. 3b, entry 7)41. Other bismuth(I) complexes and different redox couples tested led to decreased yields or inactivity, thus highlighting the importance of the finely tuned redox properties of 1 (Supplementary Information).
This methodology led us to the assembly of a wide variety of products containing an aminal- or a hemiaminal-ether structural motif, which would be challenging to construct from the parent halide (Table 1). RAEs of readily available natural and non-natural α-amino acids were investigated (either fully protected or with free N–H bonds) as electrophilic partners. C–N coupling products derived from proline (26), phenylalanine (29), valine (30 and 34), leucine (35), glutamic acid (36) or pipecolic acid (32 and 33) were successfully obtained in good to excellent yields. Synthetically relevant N-heterocycles bearing free N‒H bonds were evaluated. Using proline-derived RAE 22, the corresponding C–N products of benzimidazoles (26, 28 and 45), triazole (37), imidazoles (38, 44 and 46) and pyrazoles (32, 33 and 40–43) were obtained.
Table 1 Scope of the C–N coupling reactionNon-symmetrical heterocycles such as benzotriazole (to give 39) could also be accommodated, providing the product in a 5:1 N1/N2 ratio of regioisomers. A range of functional groups with different electronic properties were also tolerated (to give 43, 46 and 50). Since the radical process is orthogonal to classical transition-metal-catalysed cross-coupling reactions, different heteroaryl halides (to give 40–42) and heteroaryl boronic esters (to give 32 and 33) could be well tolerated. The strategy was successfully applied in the modification of bioactive molecules, such as theophylline (to give 47, 57%, single regioisomer). The successful coupling using thiabendazole (to give 48, 61%) provides another illustrative example of the orthogonal reactivity to transition metals, as the Lewis-basic sites on both starting material and product could inhibit catalysis by binding to a metal centre. Interestingly, carbamate-like N‒H bonds could also be accommodated as demonstrated by the preparation of 49, a hydroxylated analogue of riluzole. In the absence of external nucleophiles, the product of decarboxylative amination via formal CO2 extrusion was obtained. For this process, both α-amino RAEs and α-oxo RAEs reacted, giving hemiaminal-ether structures such as 50 and 51 in good yields. Overall, this strategy is complementary to the photochemical protocol reported by Fu and co-workers30, allowing the use of α-heteroatom RAEs instead of unbiased alkyl substrates.
To shed light on the mechanism, we monitored the catalytic reaction of α-amino RAE 22 by NMR with 10 mol% of 1 at −40 °C, using DMF-d7 as the solvent. In this scenario, the pair of rotamers of α-amino alkyl-bismuth(III) intermediate 23 accumulated upon consumption of the RAE, coexisting with bismuthinidene 1. It is important to mention that complex 23 was characterized by reaction of 1 with 22 in a separate stoichiometric experiment (see Supplementary Information for details). The accumulated 23 decays into 1 after 1 h at −20 °C (Fig. 4, bottom, Bi(I/II/III) pathway). However, we observed that the consumption of RAE 22 to give decarboxylative-amination product 24 occurs at a higher rate than that of the former process (see Supplementary Information for details of kinetic analysis). Thus, an alternative pathway should be considered in which the corresponding in-cage radical pair reacts directly through SET, leading to the iminium cation upon regeneration of Bi(I) (Fig. 4, bottom, Bi(I/II) pathway)46. Alternatively, radical recombination of the aforementioned radical pair leads to some accumulation of 23, which eventually collapses into the reaction product (24) and 1. Overall, the radical oxidative addition appears to be the rate-limiting step of the dominant pathway, as suggested by the continuous presence of 1 throughout the entire course of the reaction. Importantly, low-temperature EPR-spectroscopy analysis allowed us to detect an intense single-line signal, in agreement with the presence of the corresponding α-amino alkyl-radical fragment. This strong EPR signal was observed even in the dark. This is consistent with the fact that the Bi(I/II) pair can promote this reactivity in the absence of external light irradiation.
留言 (0)