
記住我



Cell death is an essential process of life and means the end of cell life. In the past, the Nomenclature Committee on Cell Death (NCCD) has developed guidelines to define and explain cell death from morphological, biochemical, and functional perspectives (1). As the field of cell death continues to evolve and new signaling pathways are discovered, NCCD adjusts how different cell deaths are defined and provides an updated classification of cell death patterns (1). Currently, cell death can be classified into accidental cell death (ACD) and regulatory cell death (RCD) based on functional aspects (2). ACD can be triggered by unexpected attacks and injuries, such as exposure to extreme physicochemical or mechanical stressors, which would overwhelm control mechanisms.
In contrast, RCD is a regulated signaling cascade executed by specific effector molecules and carries unique biochemical, functional, and immunological consequences (3). RCD can be divided into several subroutines based on its molecular characteristics, several of which have clear physiological significance (e.g., necroptosis and pyroptosis). In contrast, others may be cellular responses to specific toxins and do not reflect normal physiology(e.g., ferroptosis, entotic cell death, netotic cell death, parthanatos, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis, and oxeiptosis) (3). Ferroptosis is a form of RCD, a concept first proposed by Dixon and colleagues in 2012 (4).
Ferroptosis is cell death that depends on iron metabolism disorder and is characterized by intracellular lipid peroxides (LPO) accumulation. Specific morphological changes in ferroptosis include mitochondrial atrophy and increased mitochondrial membrane density (5). Biochemically, ferroptosis is characterized by the depletion of glutathione (GSH), the decrease of glutathione peroxidase 4 (GPX4) activity, and the accumulation of Fe2+ and LPO, thus destroying the typical structure and function of cells (4, 6).
Diabetic retinopathy is one of the significant complications of diabetes mellitus, which can cause visual impairment and blindness. The severity is divided into non-proliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR). The earliest morphological sign of NPDR is the formation of retinal microaneurysms. With the further progress of the diseases, blot hemorrhage, hard exudation, and cotton wool spots in the retina were observed (7). PDR is characterized by neovascularization, fibrovascular proliferation, and leakage of retinal capillaries. Vitreous hemorrhage and tractional retinal detachment develop as the disease progresses (8). The occurrence of DR may be related to the abnormal regulation of many biochemical pathways caused by elevated blood glucose levels. This abnormal regulation ultimately leads to superoxide production and oxidative stress burden in the retinal tissue. Meanwhile, inflammation, neovascularization, and disruption of the blood-retinal barrier are also involved in developing DR (9, 10).
Ferroptosis may be associated with some diseases, such as cancer, brain diseases, immune system diseases, and ischemia/reperfusion. Pathological mechanisms such as oxidative stress, inflammation, and neovascularization are associated with the occurrence and development of these diseases. DR is a metabolic disorder with a similar pathology to these diseases, so DR may also be associated with iron-dependent cell death. However, comprehensive information on the pathophysiology of ferroptosis in DR needs to be comprehensive. In this review, we focus on the recent research development of ferroptosis and its pathological mechanism in DR. Meanwhile, and this paper reviews possible therapeutic targets for ferroptosis in DR. In addition, some questions related to this research field and future research ideas are proposed.
2 Mechanisms of ferroptosisFerroptosis is a type of RCD with unique morphology and pathological mechanisms. The cystine/Glutamate Antiporter Pathway, System Xc-, is considered to be a critical signaling pathway associated with iron-dependent cell death (5). Lipid peroxidation and iron metabolism are also important events in ferroptosis (11). Meanwhile, GPX4 is a crucial enzyme that degrades peroxides and inhibits ferroptosis. Physiologically, Fe3+ enters cells through the transferrin receptor (TFR) and is converted to Fe2+, an essential substance for cellular energy metabolism. However, excess Fe2+ reduces oxygen to form superoxide radicals and causes lipid peroxidation (12). Therefore, the inactivation of GPX4 and iron metabolism disorders causes intracellular LPO accumulation (13). Furthermore, studies have suggested that p53, a well-known oncogene, is associated with ferroptosis in tumor cells (14).
2.1 System Xc-A cystine/glutamate antiporter, System Xc-, is an amino acid anti-transporter distributed in phospholipid bilayers (15). System Xc- comprises a twelve-channel transmembrane transporter protein SLC7A11 coupled to a single-channel transmembrane regulator protein SLC3A2 via a disulfide bond (16). It imports cystine and exports glutamate, which is critical in maintaining intracellular GSH levels and extracellular cystine/cysteine redox balance (17). The cystine taken up is reduced to cysteine in cells, which is involved in the synthesis of GSH.
2.2 GPX4 and GSHAmong the members of the GPX family, GPX4 plays a vital role in ferroptosis, mainly by inhibiting the formation of LPO. GSH acts as an essential cofactor of GPX4, which reduces hydrogen peroxide and organic peroxides to water and corresponding alcohols, thereby eliminating cytotoxicity (11). Therefore, inhibition of GPX4 activity leads to the accumulation of LPO, a critical factor in ferroptosis (18). It was discovered that removing GPX4 in mouse tissues leads to ferroptosis in the corresponding cells (19). Complete deletion of the GPX4 gene has been reported to be lethal in the embryo, resulting in the death of specific cell types in particular tissues (20, 21).
2.3 Lipid peroxidationBoth non-enzymatic and enzymatic methods can induce lipid peroxidation, which is crucial in cell ferroptosis (22). Non-enzymatic radical chain reactions are related to Fenton chemistry and produce highly toxic hydroxyl and peroxy radicals. Enzyme-dependent reaction processes involve iron-containing enzymes such as lipoxygenase and arachidonate lipoxygenase (ALOX) (23, 24). Free polyunsaturated fatty acids(PUFAs) are esterified into membrane phospholipids and oxidized as substrates for synthesizing lipid signal transducers (25). PUFA-containing phospholipids (PUFA-PLS) lipid radicals extract protons from adjacent polyunsaturated fatty acids and trigger a new round of lipid oxidation, further spreading oxidative damage from one lipid to another and accelerating the production of LPO (11). Therefore, intracellular lipid peroxidation is regulated by the content and location of PUFA. The exact mechanism by which lipid peroxidation induces ferroptosis and the precise location in cells where this phenomenon occurs are being investigated.
2.4 Iron metabolismAs a trace element, iron plays an essential role in maintaining the normal physiological metabolism of the body. Fe2+ absorbed by the intestine can be oxidized to Fe3+ by ceruloplasmin. Fe3+ binds to transferrin(TF) and forms a complex with transferrin receptors 1(TFR1)on the cell membrane before entering the cell (26). Fe3+ is reduced to Fe2+ in the cell, some are stored in unstable iron pools, and others are bound to ferritin. The iron in ferritin is released by lysosomal degradation, increasing intracellular iron concentration (27). Fe2+ reduces oxygen to form superoxide radicals, which induce lipid peroxidation and thus lead to cell ferroptosis (28).
2.5 P53Recent studies have shown that tumor suppressor p53-induced ferroptosis can be a tumor suppressive mechanism. P53 was first reported to induce cell ferroptosis through transcriptional repression of SLC7A11, a specific system-Xc subunit (29). After combining with p53-specific binding proteins, p53 acts on SLC7A11 and inhibits its expression in the promoter region, resulting in enhanced sensitivity of cancer cells to ferroptosis inducers (29). In addition to system Xc-, p53 upregulates its downstream recombinant spermidine/spermine/spermine N1-acetyltransferase 1 (SAT1) to induce ferroptosis (30). In human cancers, wild-type p53 is degraded by the oncogene E3 ubiquitin-protein ligase MDM2. Thus, inhibition of MDM2-dependent proteasomes may play a role in p53-induced ferroptosis, providing novel cancer treatment strategies (27).
With the increased research on iron death, we found that it is involved in various pathophysiological processes in cells. However, ferroptosis involves complex metabolic mechanisms, and a single inhibitor is insufficient evidence (31). It is to be explored how to avoid the interference of other mechanisms in the demonstration of cell ferroptosis. In addition, the specific molecular mechanisms and pathways of ferroptosis in disease remain to be explored.
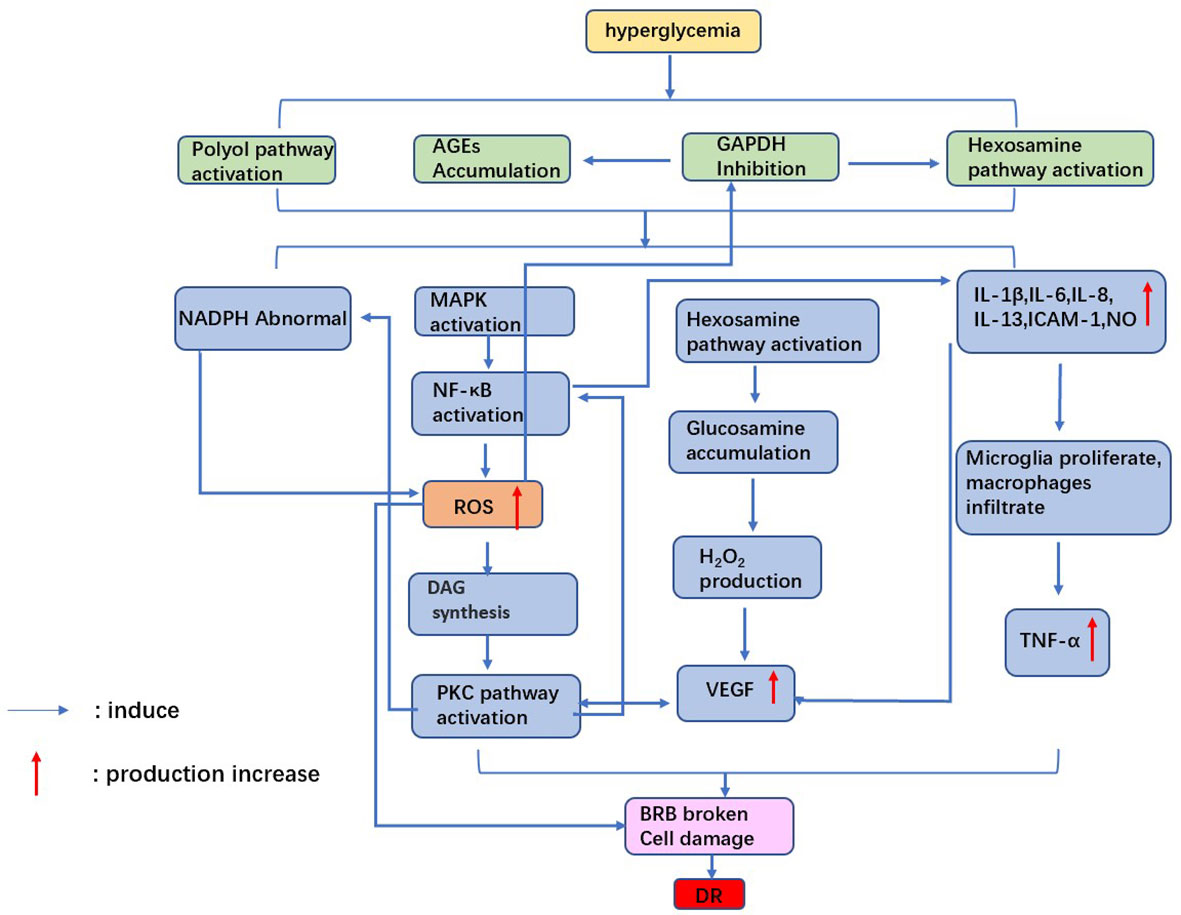
3 Diabetic retinopathyAlthough many of the underlying pathogenesis of DR are not fully understood, oxidative stress, inflammation, neovascularization, and disruption of the blood-retinal barrier are involved in the development of DR (7, 8). This review will describe the known pathological mechanisms (Figure 1).
Figure 1 Mechanism of diabetic retinopathy. Hyperglycemia leads to oxidative stress and inflammatory factor release, which damages retinal cells. the accumulation of VEGF causes neovascularization. Ultimately, the blood-retinal barrier is broken, causing diabetic retinopathy.
3.1 Oxidative stressOxidative stress generated by hyperglycemia-induced mitochondrial superoxide excess is one of the pathological mechanisms of DR (32). Massive accumulation of ROS leads to mitochondrial dysfunction, apoptosis, local Inflammation, and microvascular dysfunction, significantly affecting all DR pathogenesis (33). Under hyperglycemia, glucose binds to large molecules such as proteins, lipids, and amino acids in nucleic acids, forming AGEs (34). Accumulation of AGEs can cause retinal vascular obstruction, resulting in retinal ischemia and activation of intracellular signaling pathways such as mitogen-activated protein kinase (MAPK) and nuclear transcription factors-κB(NF-κB). These processes trigger cellular hypoxia, ROS production, and cell damage (35). Furthermore, high levels of ROS inhibit glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity, leading to a flow of glycolytic products into the hexosamine pathway (36). Glucosamine production from activated hexosamine increases hydrogen peroxide production, further leading to cell oxidative damage and enhanced vascular permeability. Meanwhile, inhibition of GAPDH will induce the activity of the AGE pathway, further enhancing oxidative stress in retinal cells (37).
Protein kinase C (PKC) is a serine/threonine kinase involved in signal transduction that responds to the stimulation of specific hormones, neurons, and growth factors (38). ROS accumulation and diacylglycerol (DAG) synthesis under hyperglycemia leads to activation of the PKC pathway. It was found that activation of the PKC pathway could induce endothelial cell damage through increased endothelial cell permeability, altered NO bioavailability, and reduced prostaglandin production (39). In addition to the above pathways, hyperglycemia causes abnormalities in polyol pathways, resulting in reduced NADPH availability and increased cellular sensitivity to oxidative stress (40). On the one hand, retinal cells are exposed to oxidative damage induced by high glucose. On the other hand, retinal antioxidant capacity is reduced under high glucose. Therefore, the combined effect of both factors causes retinal damage.
3.2 InflammationChronic Inflammation is associated with developing diabetes and its complications (41, 42). For example, hyperglycemia causes cellular damage by inducing microglia to secrete cytokines and other inflammatory molecules and disrupting the balance of the cellular environment (43). Shelton et al. (44) found that IL-1β, IL-6, IL-8, IL-13, ICAM-1, and NO increased in Muller cells and retinal vascular endothelial cells during DR, which confirmed that inflammatory factors were related to the development of DR.
In addition, increased diacylglycerol (DAG) production via the glycolytic pathway activates PKC under hyperglycemia (45). Activation of PKC drives overexpression of NADPH oxidase and NF-κB in cells, thereby exacerbating DR-related oxidative stress and inflammatory responses (46). It was found that the plasma and vitreous concentrations of TNF-α were increased in patients with diabetes, and the plasma TNF-α concentration was related to the severity of DR (47).In diabetic rats, the vitreous TNF-α concentration was related to the increased permeability of BRB (48). Therefore, the chronic inflammatory response caused by hyperglycemia creates long-term damage to retinal cells, affecting the repair ability of cells and promoting DR development.
3.3 Vascular endothelial growth factor and neovascularizationNeovascularization is one of the critical features of PDR. Occlusion of retinal microvessels can lead to retinal ischemia and neovascularization. This process is accompanied by the release of vascular endothelial growth factor (VEGF), a glycoprotein that increases retinal vascular permeability and promotes neovascularization. There are four subtypes of VEGF, including VEGF-121, -165, -189, and -206. VEGF-165 has proven to be most closely associated with DR pathology (49).
VEGF destroys retinal cells mainly through the following factors. Firstly, VEGF induces ICAM-1 expression and leukocyte adhesion in the retina, resulting in BRB rupture, capillary nonperfusion, and endothelial cell injury. Secondly, VEGF promotes tight junction disassembly, vascular permeability, and BRB disruption by inducing PKC activation and phosphorylation of tight junction proteins (50). Activation of PKC-B2 isoforms is reported to be critical in the mechanism of VEGF-induced DR neovascularization (51). Finally, VEGF induces the expression of serine proteinases, tissue fibrinogen activators, and metalloproteinases while significantly reducing tissue inhibitors of metalloproteinases. These factors lead to abnormal retinal angiogenesis and proliferation (52).
3.4 Blood-retinal barrier impairmentThe outer blood-retinal barrier(oBRB) comprises the choroid, Bruch’s membrane, and RPE. The inner blood-retinal barrier (iBRB) comprises the retinal endothelium and pericytes, essential for maintaining retinal tissue integrity and normal retinal function (53). One of the causes of retinal edema and macular edema in diabetic patients is BRB disruption and fluid accumulation in the retinal space. Injury of BRB under hyperglycemia may be related to the following factors.
On the one hand, Inflammation caused by hyperglycemia plays an essential role in destroying retinal integrity. It was found that IL-6 levels in retinal tissue were elevated in an animal model of diabetes, which would upregulate levels of mRNA encoding microglia chemokines and result in microglia proliferation (54). TNF-α production and secretion by IL-6-treated microglia reduced zonula occludens-1(ZO-1) in RPE cells and disrupted the tight junction complex, thereby affecting the integrity of the oBRB. In addition, IL-1β and IL-8 also upregulated the expression of VEGF in diabetic animal models, which resulted in a substantial reduction in pericytes, capillary deterioration, and breakdown of BRB.
On the other hand, the integrity of RPE cells is also closely related to the electrical activity of the cell membrane. Na+/k+-ATPase is an essential source of energy for RPE cells. It uses the energy of ATP to pump 3 Na+ out of the cell and every 2 K+ into it. Na+/k+ - ATPase activity is significantly reduced in a hyperglycemic environment, increasing intracellular osmotic pressure, retinal cell edema, and retinal barrier (55).
Retinal tissue comprises multiple cells, so diabetic retinopathy involves pathological changes in different cells, which is complicated and elongated. Based on the continuous development of clinicopathological and experimental data over the past years, a broader view of diabetic retinopathy and its progress is now available (32). However, treating diabetic retinopathy in clinical practice mainly focuses on the intermediate and late stages. More in-depth studies on the pathogenesis are still needed for early treatment and prevention.
4 Research progress into ferroptosis in diabetic retinopathy4.1 Reactive oxygen speciesOxidative stress is associated with developing several neuroretinal degenerative diseases, such as AMD and DR. Oxidative stress is cell damage caused by an imbalance between oxidants and antioxidants. It causes excessive accumulation of ROS and damage to cellular proteins, lipids, and DNA. On the one hand, hyperglycemia disrupts mitochondrial homeostasis to produce massive amounts of ROS, causing local tissue and cellular hypoxia. Long-term chronic hypoxia, in turn, aggravates oxidative stress damage to cells. Dimethyloxallyl Glycine (DMOG) stabilizes hypoxia-inducible factors and promotes cellular adaptation to hypoxia (56). Sodium iodate (SIO) and DMOG-treated ARPE-19 increase superoxide dismutase activity and Fe3+ levels, intensifying the Fenton reaction (56).
On the other hand, ROS reacts with phospholipids in biological membranes, enzymes, and side chains of polyunsaturated fatty acids and nucleic acids to form LPO such as malonaldehyde (MDA) and 4-hydroxynonenal (HNE) through lipid peroxidation. Eventually, the fluidity and permeability of cell membranes change, and cell structure and function are affected. Zhou Jing et al. discovered that the ROS scavenger, n-acetyl-l-cysteine (NAC), inhibited high glucose-induced ferroptosis in RPE cells (ARPE-19) after the elimination of ROS (45). Therefore, oxidative stress was shown to be involved in cell ferroptosis under hyperglycemia.
As an antioxidant, nuclear factor erythroid 2-related factor 2 (NRF2) is a redox-sensitive essential leucine zipper region transcription factor, which plays a crucial role in protecting the retinal vascular system from ROS-related damage (35). Nrf2 enters the nucleus by binding to genes associated with multiple antioxidant reaction elements, thereby promoting the expression of target genes such as GPX, heme oxidase-1 (HO-1), and glutamine-cysteine ligase catalytic subunit (GCLC) (57). The activity of NRF2-related antioxidant enzymes decreased in an in vitro model of diabetes. Therefore, NRF2 depletion leads to uninhibited NOX2 activation and triggers ROS accumulation (58), which causes cell damage in diabetes models. Several studies have identified some factors associated with the regulation of Nrf2. For example, Silencing information regulator 1(Sirt1), a nicotinamide adenine nucleotide (NAD) -dependent protein deacetylase, has been shown to regulate Nrf2 expression and activity positively, thereby inhibiting cell ferroptosis (59). Ras-selection-lethal compound 3(RSL3) inhibits the activity of GPX4, which leads to the accumulation of ROS and reduces the antioxidant capacity of cells (13). However, intraperitoneal injection of RSL3 increased Nrf2 expression in mouse microglia, thus partially inhibiting the production of inflammatory cytokines and, to some extent increasing the resistance of cells to ferroptosis (60).
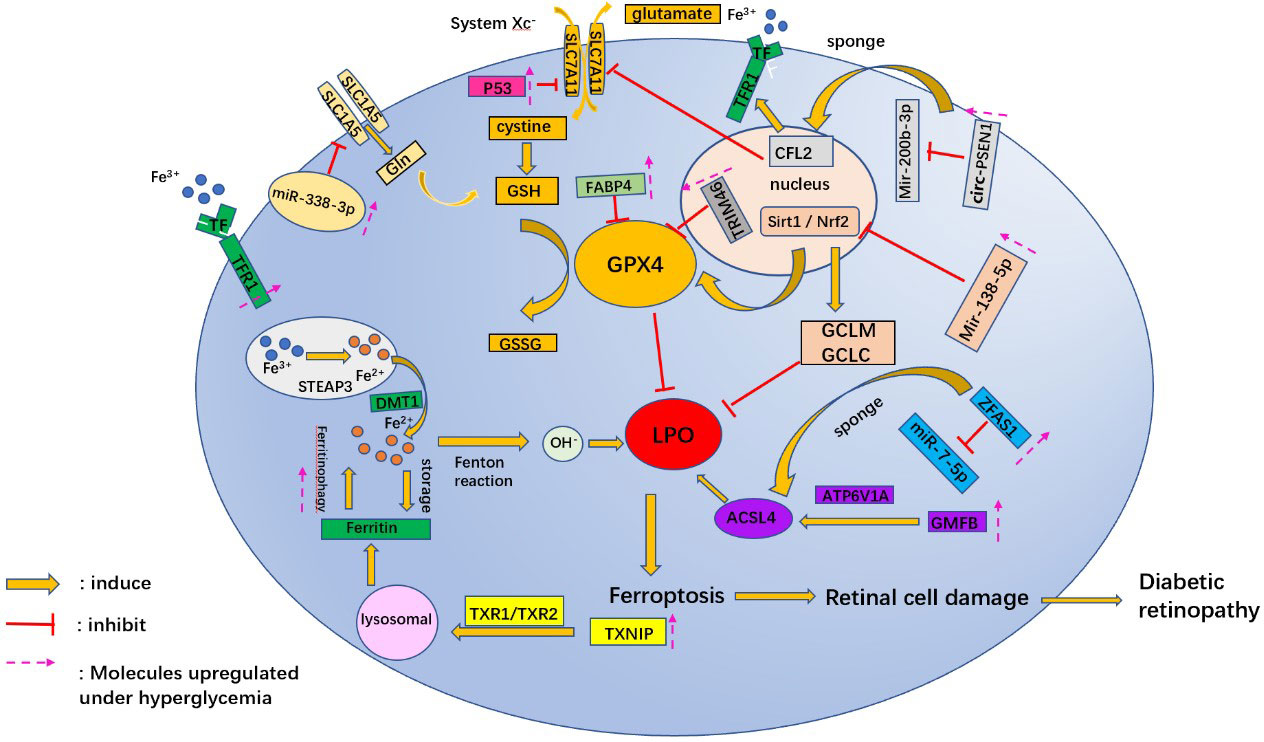
Recent studies have also identified target miRNAs associated with oxidative stress in DR (61), such as miR-338-3p (62). miR-338-3p targets the 3 ‘untranslated region (3’UTR) of SLC1A5 for inhibition and degradation. Hyperglycemia triggers ferroptosis in ARPE-19 cells by regulating the Mir-338-3p/SLC1A5 axis. The main mechanism is that overexpression of Mir-338-3p and inhibition of SLC1A5 promote intracellular ROS, total iron, and ferrous iron levels (45) (Figure 2).
Figure 2 A schematic picture shows the potential mechanism associated with ferroptosis in diabetic retinopathy. Gln, glutamine; GSH, L-Glutathione; GPX4, glutathione peroxidase 4; GSSG, Oxidized glutathione; TRIM46, tripartite motif 46; CFL2, Cofilin2; Mir, micro RNA; circ-PSEN1, circular RNA- Presenilin 1; sirt1, Sirtuins 1; Nrf2, nuclear factor-erythroid 2-related factor; GCLM, glutamate-cysteine ligase modifier subunit; GCLC, glutamate-cysteine ligase catalytic subunit; FABP4, fatty acid binding protein 4; LPO, lipid peroxides; GMFB, glial maturation factor B; ATP6V1A, 1V-type proton ATPase catalytic subunit A; ACSL4, acyl-CoA synthetase long-chain family member 4; TF, transferrin; TFR, transferrin receptor; STEAP3, STEAP family member 3; DMT1, Divalent metal cation transporter 1; TXNIP, thioredoxin-interacting protein; TrxR1, thioredoxin reductase 1; ZFAS1, zinc finger antisense 1.
4.2 GPX4 and GSHGPX protects cells from oxidative stress damage and inhibits cell ferroptosis. The outer segments of photoreceptors are the site of high concentrations of PUFAs and are vulnerable to oxidative stress (63). Knockdown of GPX4 isoforms completely in mouse photoreceptors leads to an early degenerative phenotype, which reveals the importance of GPX4 for photoreceptor survival (21). Cultivation of RPE cells in a GSH-depleted medium resulted in decreased GPX4 expression and increased LPO production. In addition, GSH depletion induced premature senescence in RPE cells, as evidenced by an increased percentage of senescence-associated β-galactosidase-positive cells, increased senescence-associated heterochromatin foci, and cell cycle arrest at the G1 phase. Therefore, GSH depletion leads to RPE cell degeneration and ferroptosis (64).
miRNAs regulate post-transcriptional gene expression and cell ferroptosis by affecting GPX4 expression in hyperglycemic conditions. Overexpression of Mir-138-5p inhibited the activity of the Sirt1/Nrf2 pathway in a hyperglycemic environment and reduced the antioxidant capacity of RPE cells. The primary mechanism was to inhibit downstream molecules of Sirt1/Nrf2, such as GPX4, glutamine-cysteine ligase modified subunit (GCLM), and glutamine-cysteine ligase catalytic subunit (GCLC) proteins (65). Tang X et al. (66) discovered that Astragaloside IV (AS-IV) attenuated ferroptosis in RPE cells by inhibiting Mir-138-5p expression. In addition to miRNA, lncRNAs are also involved in regulating gene expression of ferroptosis in retinal cells. lncRNA zinc finger antisense 1 (ZFAS1) expression was upregulated in hRECs cultured with high glucose. ZFAS1 may promote the expression of downstream mRNA ACSL4 and reduce the level of GPX4 by competitive binding to miR-7-5p under a high glucose environment (66).
High glucose levels have been found to promote the activation of the oncogene p53 (67). To further determine whether the p53-System Xc–GSH axis is involved in HG- and IL-1β-induced iron death in endothelial cells, Luo EF et al. transiently transfected HUVECs with p53 siRNA or NC siRNA, followed by treatment with NG, HG, and IL-1β. The results revealed that p53 siRNA significantly reduced the expression of HG- and IL-1β-induced xCT and GSH (68).
Ferrostatin-1 was the first ferroptosis inhibitor developed to inhibit LPO by scavenging the initiating alkoxy radical and other rearrangement products (69). It was found that Ferrostatin 1 significantly reduced the expression of MDA, one of the lipid peroxidation products, as well as increased the proteins of SLC7A11 and GPX4 in photoreceptor cells (54). Besides), Inhibitors (e.g., elastin (4), sorafenib (70), or sulfasalazine (71))that act on SLC7A11 lead to depletion of GSH and inactivation of GPX4, resulting in LPO accumulation and cell ferroptosis.
4.3 Lipid peroxidationAs is known, one of the critical features of ferroptosis is the accumulation of LPO. High glucose leads to the destruction of the blood-brain barrier and the release of many divalent iron ions, which causes the overproduction of ROS in the Fenton reaction and oxidizes cell membrane lipids (23). FABP4 is a small molecule protein vital in maintaining glucose and lipid homeostasis. FABP4 is highly expressed in diabetic complications. Inhibition of FABP4 ameliorates high glucose-induced glomerular apoptosis and increases serum insulin concentration, thereby attenuating the progression of diabetes-relevant diseases. Therefore, FABP4 participates in and regulates the development of diabetes-related diseases (72). Fan X et al. used the FABP4 inhibitor BMS309403 to enhance the activity of GPX4 and reduce lipid peroxidation, thereby protecting diabetic mouse retinal cells from oxidative stress (73).
Other molecules, such as TRIM46, are also involved in regulating lipid Peroxidation in hyperglycemia. TRIM46, a gene localized on chromosome 1q21, could mediate the ubiquitination of GPX4. Overexpression of TRIM46 in high-glucose treated human retinal microvascular endothelial cells inhibits GPX4 activity and increases ROS and MDA, whereas TRIM46 knockdown produces the opposite effect (74). Furthermore, TRIM46 overexpression aggravates the damage of Human retinal microvascular endothelial cells (HRCECs) in high glucose conditions by promoting the degradation of IκBα and the activation of the NF-κB pathway (75).
In addition, circRNAs affect cell ferroptosis in DR by regulating the expression of miRNAs (76). For example, the knockdown of circRNA PSEN1 ameliorates high glucose-induced ferroptosis in RPE cells via the Mir-200b-3p/cofilin-2(CFL2)axis. MiR-200b-3p is sponged by circ-PSEN1, which leads to enhanced expression of CFL2. Overexpression of CFL2 results in repression of mRNA and protein expression of GPX4 and SLC7A11, while expression of TFR1 is promoted (77). These factors cause an increase in intracellular Fe2+ and LPO, which leads to cell ferroptosis.
4.4 Mitochondrial and lysosomal dysfunctionMitochondria are organelles that provide energy for various physiological activities of the cell. Lysosomes are rich in hydrolytic enzymes and perform intracellular digestive functions. Mitochondrial and lysosomal dysfunction can induce cell ferroptosis. Mitochondria consume large amounts of glucose and oxygen to produce ATP. ATP is necessary to satisfy the high energy demand required for retinal (78) so a significant reduction in ATP levels can affect visual function. However, many reactive oxygen radicals accompanying ATP production can damage mitochondrial membranes, proteins, and mitochondrial DNA (79). ROS destroy mitochondria in a high glucose environment, and damaged mitochondria produce less ATP but continue to produce ROS (80). Under hyperglycemia, thioredoxin-interacting protein (TXNIP) expression is enhanced in ARPE-19 cells (81). It induces autophagy flux, mitochondrial dysfunction, and lysosomal instability by binding to and inhibiting thioredoxin reductases 1 and 2 (TrxR1 and TrxR2) (82). A large amount of Fe2+ is released from the lysosome during this process. The released Fe2+ reacts with hydrogen peroxide to produce many hydroxyl radicals, resulting in lipid peroxidation of lysosomes, mitochondria, and plasma membranes. This process damages the organelles and plasma membrane of the cells (83). Several Dihydrolipoic acids (ALA) derivatives, including Dihydrolipoic acid, are known to protect mitochondria from oxidative stress-induced damage by upregulating the anti-inflammatory and antioxidant defense systems of cells (84).
RPE cells phagocytose the exfoliated outer segment membrane discs of photoreceptors to provide nutrients to the retinal neuroepithelial layer. High glucose causes mitochondrial damage, lysosomal overload, and reduced protein digestion ability in RPE cells. Undigested lipofuscin is deposited in the Bruch membrane, thereby damaging the integrity of the RPE barrier function. Therefore, removing damaged mitochondria by mitochondrial phagocytosis reduces intracellular ROS and protects cells from oxidative stress damage. Thangal Yumnamcha et al. (82) reduced the incidence of ferroptosis in RPE cells by combining SS31 (mitochondrial antioxidant), ammonia oxidant (inhibition of TBK1 and normalization of mitochondrial phagocytic flux), and tranilast (inhibition of NLRP3) to inhibit mitochondrial dysfunction. Thus, the three-drug combination may be helpful in the treatment of mitochondria-associated neurodegenerative diseases, such as DR.
In hyperglycemia, the function of lysosomes is affected by several acidic proteins. It was found that the concentration of glial maturation factor-β (GMFB) in the vitreous cavity of diabetic rats was significantly increased (65). GMFB affects the degradation process of autophagolysosome through ATP6V1A translocation and induces the accumulation of ACSL4 (partner-mediated autophagy substrate), ultimately affecting normal cell metabolism and destroying the physiological functions of the retina (65).
5 Conclusion and outlookFerroptosis has received more research and attention as a mode of RCD. This paper provides a more comprehensive description of the molecules and pathways associated with iron toxicity. It mainly includes abnormal regulation of System Xc-, depletion of GSH, and inactivation of GPX4, as well as abnormal intracellular iron metabolism, accumulation of LPO, and regulation of P53. The molecules and pathways associated with ferroptosis are complex and require further investigation. There are few studies on the relationship between ferroptosis and ocular diseases. There is no comprehensive review of ferroptosis in the pathogenesis of DR. Therefore, in this paper, the existing mechanisms of ferroptosis associated with the development of DR are described to understand the pathology of DR further (Tables 1, 2).
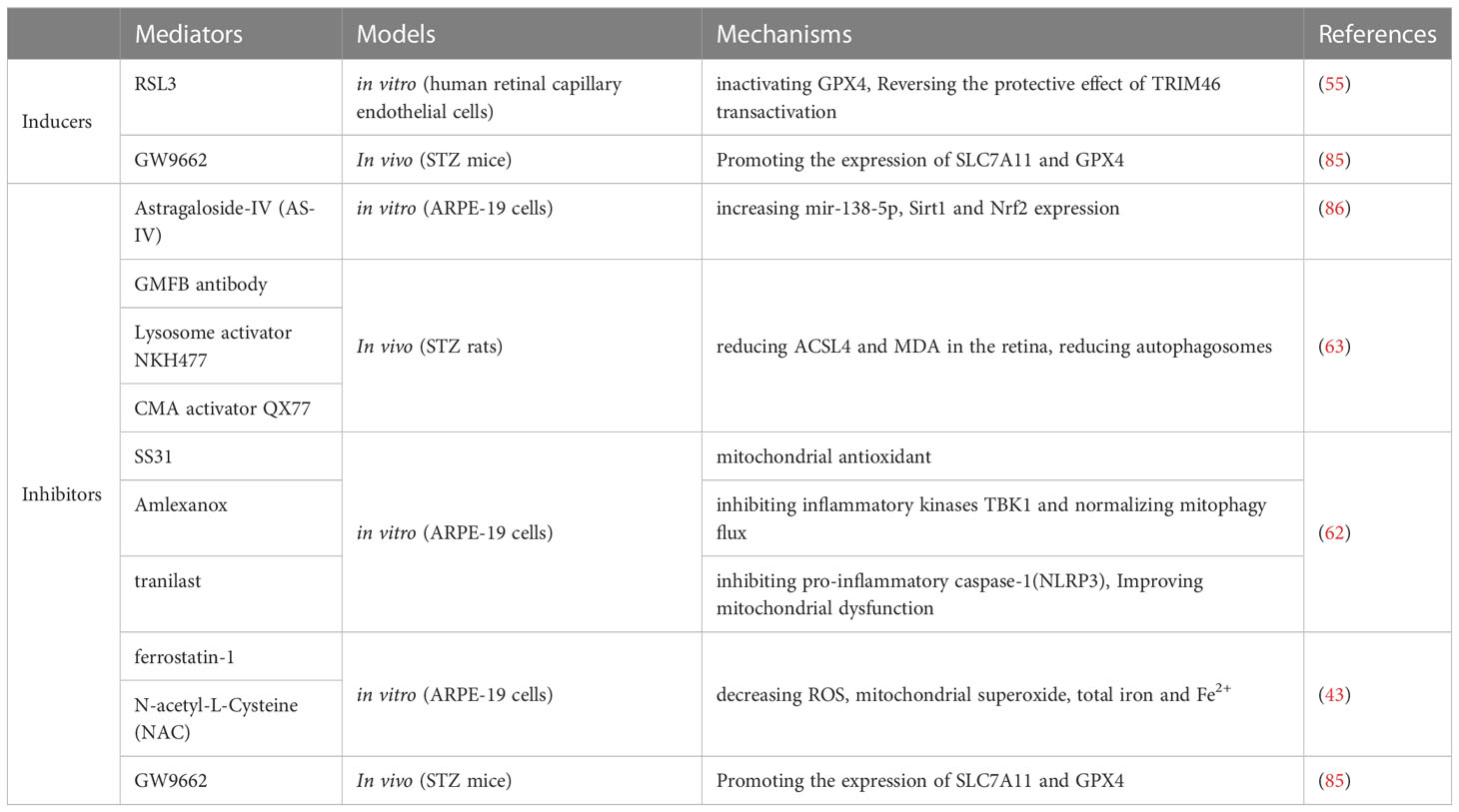
Table 1 Mediators of ferroptosis in diabetic retinopathy.
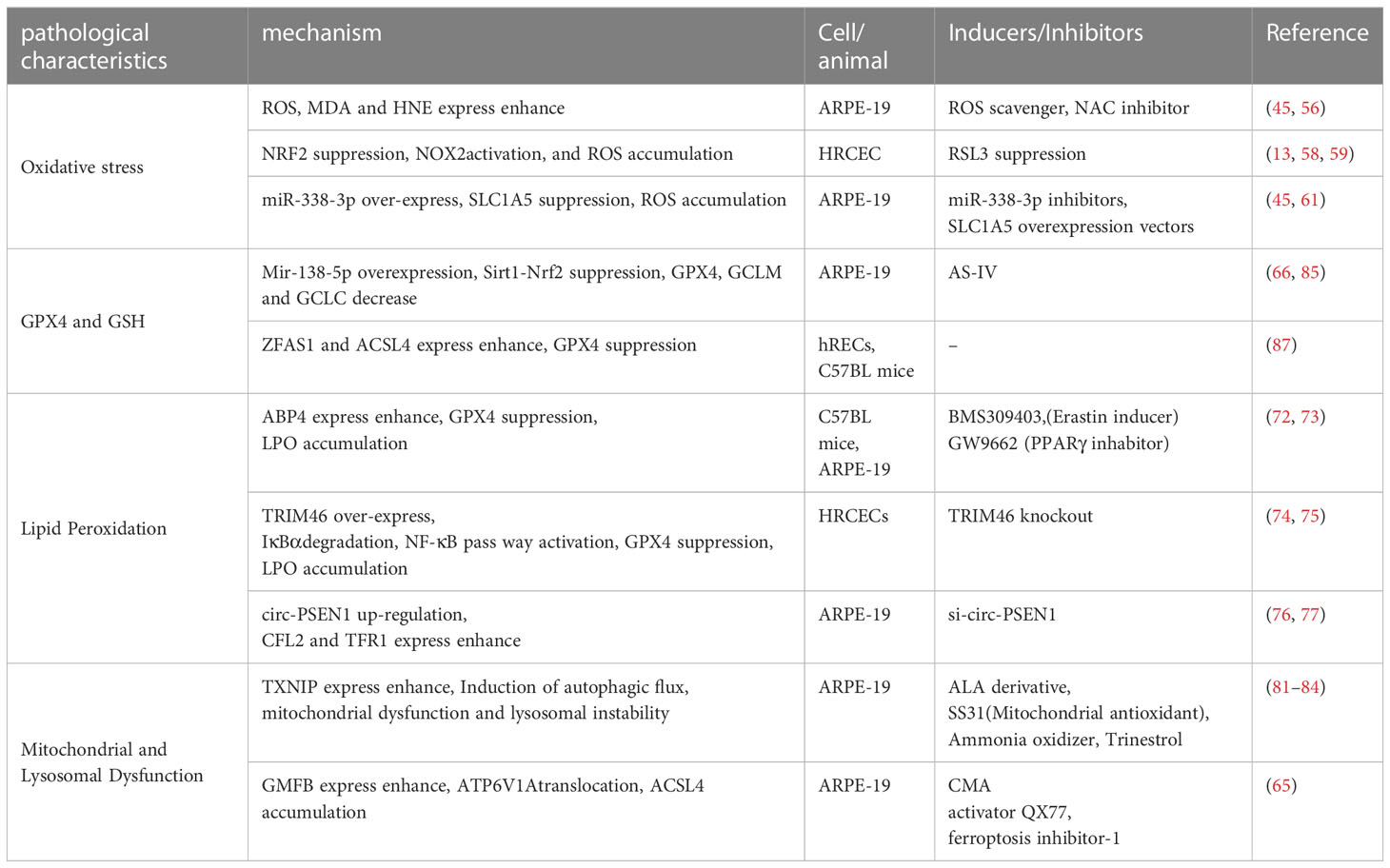
Table 2 Pathogenesis of ferroptosis in diabetic retinopathy.
DR is a metabolism-related disease with complex pathology that seriously threatens visual function. Current treatment for DR includes control of blood glucose and blood pressure, retinal laser photocoagulation, anti-VEGF, intravitreal corticosteroids, and vitrectomy. Photocoagulation causes some damage to retinal tissue, and vitreous corticosteroids may lead to increased intraocular pressure and secondary glaucoma. Multiple vitreous injections increase the risk of intraocular infection. Moreover, vitrectomy may cause medical damage to the retina, and surgical treatment of the cause of DR cannot be performed. Therefore, more than the effects of current treatments is needed. This paper discusses the role of ferroptosis in the development of DR and complements the pathogenesis of DR, providing new ideas for treating DR.
However, current research focuses on animal and in vitro experiments. Although the relevance of ferroptosis to DR has been confirmed, whether its pathological mechanisms in animal models or in vitro are closely aligned with those in humans needs further proof. It is also unclear whether ferroptosis inhibitors suppress the development of DR in humans. Therefore, the clinical application of ferroptosis-related therapy still needs further exploration. Future research on ferroptosis will have positive significance for DR prevention, control, and treatment.
Author contributionsWH completed the manuscript. LC assisted in the completion of the manuscript. YM was responsible in the revision of articles XL assisted in the literature search for the manuscript. All authors contributed to the article and approved the submitted version.
FundingThis work was supported by Makoto Inoue expert workstation in Yunnan Province (grant no.202005AF150030).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ (2015) 22(1):58–73. doi: 10.1038/cdd.2014.137
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Ke B, Tian M, Li J, Liu B, He G. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy: ANTICANCER COMPOUNDS TARGETING CELL DEATH. Med Res Rev (2016) 36(6):983–1035. doi: 10.1002/med.21398
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16(12):1180–91. doi: 10.1038/ncb3064
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Kollias AN, Ulbig MW. Diabetic retinopathy. Deutsches Ärzteblatt international (2010) 107(5):75–83. doi: 10.3238/arztebl.2010.0075
CrossRef Full Text | Google Scholar
8. Tan Y, Fukutomi A, Sun MT, Durkin S, Gilhotra J, Chan WO. Anti-VEGF crunch syndrome in proliferative diabetic retinopathy: a review. Survey Ophthalmol (2021) 66(6):926–32. doi: 10.1016/j.survophthal.2021.03.001
CrossRef Full Text | Google Scholar
11. Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv H, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Letters (2020) 483:127–36. doi: 10.1016/j.canlet.2020.02.015
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Biophys Acta (BBA) - Gen Subjects (2012) 1820(3):188–202. doi: 10.1016/j.bbagen.2011.10.013
CrossRef Full Text | Google Scholar
14. Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, et al. Acetylation is crucial for p53-mediated ferroptosis and tumor suppression. Cell Rep (2016) 17(2):366–73. doi: 10.1016/j.celrep.2016.09.022
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane Cystine/Glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274(17):11455–8. doi: 10.1074/jbc.274.17.11455
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Bridges RJ, Natale NR, Patel SA. System xc- cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS: system xc- cystine/glutamate antiporter. Br J Pharmacol (2012) 165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA (2016) 113(34):E4966–75. doi: 10.1073/pnas.1603244113
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun (2003) 305(2):278–86. doi: 10.1016/S0006-291X(03)00734-4
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ueta T, Inoue T, Furukawa T, Tamaki Y, Nakagawa Y, Imai H, et al. Glutathione peroxidase 4 is required for maturation of photoreceptor cells. J Biol Chem (2012) 287(10):7675–82. doi: 10.1074/jbc.M111.335174
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta (BBA) - Gen Subjects (2017) 1861(8):1893–900. doi: 10.1016/j.bbagen.2017.05.019
CrossRef Full Text | Google Scholar
23. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol (2011) 31(10):2040–52. doi: 10.1128/MCB.01437-10
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Ng SW, Norwitz SG, Norwitz ER. The impact of iron overload and ferroptosis on reproductive disorders in humans: implications for preeclampsia. IJMS (2019) 20(13):3283. doi: 10.3390/ijms20133283
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Peng JJ, Song WT, Yao F, Zhang X, Peng J, Luo XJ, et al. Involvement of regulated necrosis in blinding diseases: focus on necroptosis and ferroptosis. Exp Eye Res (2020) 191:107922. doi: 10.1016/j.exer.2020.107922
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Wu MY, Yiang GT, Lai TT, Li CJ. The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid Med Cell Longevity (2018) 2018:1–12. doi: 10.1155/2018/3420187
CrossRef Full Text | Google Scholar
34. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Rodríguez ML, Pérez S, Mena-Mollá S, Desco MC, Ortega ÁL. Oxidative stress and microvascular alterations in diabetic retinopathy: future therapies. Oxid Med Cell Longevity (2019) 2019:1–18. doi: 10.1155/2019/4940825
CrossRef Full Text | Google Scholar
36. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA (2000) 97(22):12222–6. doi: 10.1073/pnas.97.22.12222
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Amadio M, Bucolo C, Leggio GM, Drago F, Govoni S, Pascale A. The PKCβ/HuR/VEGF pathway in diabetic retinopathy. Biochem Pharmacol (2010) 80(8):1230–7. doi: 10.1016/j.bcp.2010.06.033
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Whitehead M, Wickremasinghe S, Osborne A, Van Wijngaarden P, Martin KR. Diabetic retinopathy: a complex pathophysiology requiring novel therapeutic strategies. Expert Opin Biol Ther (2018) 18(12):1257–70. doi: 10.1080/14712598.2018.1545836
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Pollack RM, Donath MY, LeRoith D, Leibowitz G. Anti-inflammatory agents in the treatment of diabetes and its vascular complications. Diabetes Care (2016) 39(Supplement_2):S244–52. doi: 10.2337/dcS15-3015
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Taurone S, Spoletini M, Ralli M, Gobbi P, Artico M, Imre L, et al. Ocular mucous membrane pemphigoid: a review. Immunol Res (2019) 67(2–3):280–9. doi: 10.1007/s12026-019-09087-7
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Taurone S, Ralli M, Nebbioso M, Greco A, Artico M. Attanasio G, et al. The role of inflammation in diabetic retinopathy: a review. European Review for Medical and Pharmacological Sciences (2020) 24(20):10319–29. doi: 10.26355/eurrev_202010_23379
留言 (0)