

記住我








Liver cancer is the third most common cause of cancer-related death worldwide, and hepatocellular carcinoma (HCC) accounts for 90% of primary liver cancer (Cunha et al., 2021). HCC is one of the deadliest cancers worldwide and has become an increasingly major and growing health problem in both developing and developed countries. It has a variety of causes, including liver cirrhosis, chronic infection with hepatitis B or C virus, excessive alcohol consumption, type 2 diabetes mellitus, non-alcoholic fatty liver disease, non-alcoholic steatohepatitis, exposure to the environmental toxicant aflatoxin-B1, obesity, and metabolic syndrome (D'Souza et al., 2020; Kumar et al., 2021; Thylur et al., 2020). Currently, hepatic resection and liver transplantation are the main treatments for early-stage HCC; however, most diagnoses are already in the progressive stage, where the opportunity for surgery has passed. Early recurrence within 2 years of resection accounts for 70% of recurrent HCC cases, and the difficulty of treatment after recurrence increases (Sun et al., 2021). Furthermore, liver transplantation faces the problem of insufficient liver supply. Thus, patients with advanced HCC often choose molecular targeted therapy and immune checkpoint inhibitor therapy; however, the latter is still in its infancy. More immune cell markers must be explored to predict the response of HCC to immune checkpoint inhibitor therapy.
Tumors are complex ecosystems involving malignant cells, immune cells, stromal cells, blood vessels, and the extracellular matrix (ECM) (Anderson and Simon, 2020). Tumor cells develop and evolve into a complex and tightly linked tumor microenvironment (TME) that affects tumor growth, metastatic spread, and response to therapy (Bilotta et al., 2022). Furthermore, liver fibrosis is a pathological process necessary for the progression of chronic liver disease into cirrhosis and HCC (Roehlen et al., 2020). The profibrotic program responds to tissue injury; however, persistent injury and damage can lead to dysregulation of this process, promoting myofibroblast activity and a chronic inflammatory environment with infiltrating macrophages and lymphocytes (Wynn, 2008). Tumor-associated macrophages (TAMs) are key components of the complex TME ecology and are influenced by tumor-derived cytokines, which exhibit significant immunosuppressive effects that promote the malignancy and progression of various tumors (Zhou et al., 2021). Cancer-associated fibroblasts (CAFs) are the most abundant stromal cells in the TME and are associated with cancer progression. An increasing number of studies have focused on interactions between CAFs and different immune cells in cancer. For example, CAFs regulate macrophages by secreting cytokines that promote cancer progression (An et al., 2020). However, the mechanism by which the TME in HCC contributes to its progression remains unclear. Understanding the interactions between cell types is essential for understanding tumor development, prognosis, and treatment.
Single-cell RNA sequencing (scRNA-seq) is a powerful tool for studying cellular components and their interactions within the TME. In this study, a single-cell profile of HCC was constructed using scRNA-seq, revealing the differences in cell-type composition in the HCC TME and elucidating the interactions between tumor cells and the TME.
2 Materials and methods2.1 Data sourcesThe HCC scRNA-seq data were obtained from Gene Expression Omnibus (GEO) database GSE156625 (Platform: GPL16791) (Sharma et al., 2020). A total of 43 tumor tissue samples and 14 adjacent control samples from 14 patients with HCC were included in this study. Healthy normal liver samples, fetal liver samples, and mouse liver samples were excluded. In addition, Cancer Genome Atlas Liver Hepatocellular Carcinoma (TCGA-LIHC) data were obtained from the TCGA database (https://portal.gdc.cancer.gov/projects/TCGA-LIHC), including 374 HCC tissues with bulk RNA-seq data and the corresponding clinical information.
2.2 Construction of the single-cell atlasSingle-cell data were processed in quality control, filtering the 1% of cells with the highest and lowest feature numbers of expression, as well as cells with more than 10% mitochondrial gene expression. After quality control, the data were integrated and analyzed based on SCTransform standardization (Hafemeister and Satija, 2019). The merged data were subjected to cell clustering analysis using the default parameters of the R package Seurat (Butler et al., 2018). A single-cell atlas was constructed as described previously (Liang et al., 2022b). Cell clusters were first identified based on the FindNeighbors and FindClusters functions of the Seurat package, and then downscaled and visualized as a single-cell atlas based on the Unified Modal Approximation and Projection (UMAP) algorithm (Becht et al., 2018). Cluster-specific marker genes were identified using the FindAllMarkers function of the Seurat package, and cell types were identified based on the expression of highly specific genes in the cell clusters as well as classical cell markers.
2.3 Copy number variation analysisThe copy number variations (CNVs) in tumor cells derived from the HCC tissues were calculated using the R package inferCNV (v1.6.0; inferCNV of the Trinity CTAT Project, https://github.com/broadinstitute/inferCNV) to identify subclones of diseased cells and to infer tumor evolution.
2.4 Functional enrichment analysisTo explore biological status and functional differences between different cell subpopulations, we performed functional enrichment analysis based on the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases using the R package clusterProfiler (Yu et al., 2012). The hypergeometric test was used to compare differences, and p < 0.05 was considered statistically significant.
2.5 Construction of gene regulatory networksGene regulatory networks (GRNs) determine and maintain cell-type-specific transcriptional states; active transcription factors (TFs) and their target genes are commonly denoted as GRNs. To explore the key regulators that drive and maintain cellular state behavior, we constructed GRNs with TFs as their cores, divided them into several groups of co-expression modules, and inferred GRNs and cell states based on single-cell expression profiles with single-cell regulatory network inference and clustering (SCENIC) (Van de Sande et al., 2020). The binding patterns of the TFs were obtained from the JASPAR database (https://jaspar.genereg.net).
2.6 Pseudotime analysisDevelopment or external stimuli can change the functions of different cells in different ways, resulting in differential gene expression within the cells. Sorting these cells according to gene expression enabled further inference of cell development trajectory. In this study, the cell lineage trajectory of each cell type was inferred using Monocle3 (Trapnell et al., 2014), and the cells were projected into a low-dimensional space using UMAP.
2.7 Cell communication analysisTumor cells can interact with surrounding cells through the circulatory and lymphatic systems, thereby influencing the development and progression of cancer. Thus, to further investigate cellular interactions in the HCC microenvironment, we used the R package iTALK for cellular communication analysis. iTALK identifies high-confidence ligand–receptor interactions between cells by identifying genes that are highly or differentially expressed in cell clusters and matching these genes to a built-in ligand–receptor database (Wang et al., 2019).
2.8 Receiver operating characteristic curve and survival analysisThe R package limma (Ritchie et al., 2015) was used to normalize the TCGA-LIHC data. Receiver operating characteristic (ROC) curve analysis was performed using the R package pROC (Robin et al., 2011). In addition, to further explore the prognostic potential of highly expressed genes specific to each cell subpopulation, overall survival (OS) and recurrence-free survival (RFS) analyses of HCC were performed using the R package Survminer (https://rdocumentation.org/packages/survminer), and differences were calculated using the log-rank test with p < 0.05 considered significant.
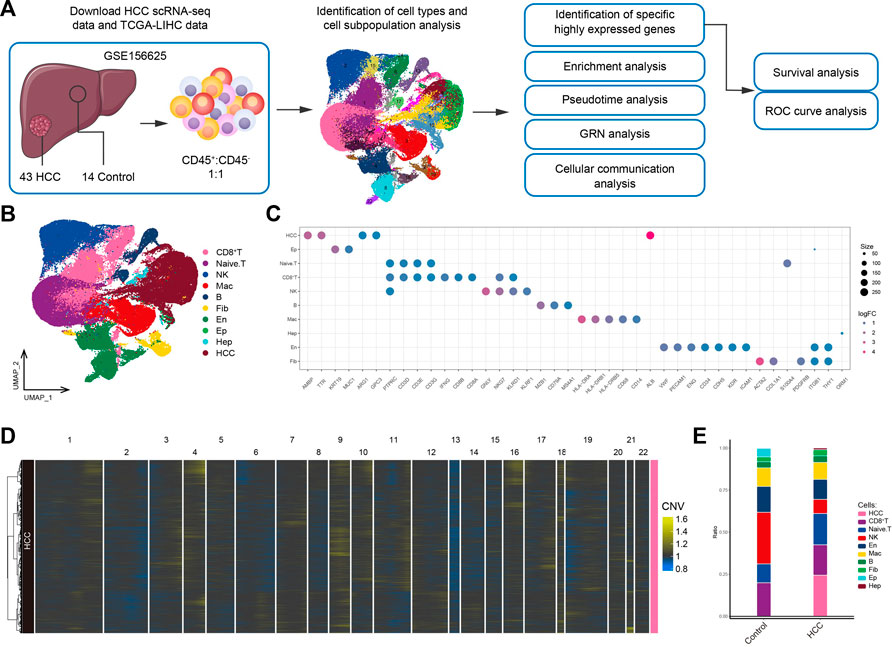
3 Results3.1 The global landscape of the HCC immune microenvironmentThe workflow of the study is shown in Figure 1A. To investigate the tumor ecological landscape in patients with primary HCC, we analyzed scRNA-seq data from GSE156625, which consisted of 43 tumor tissue samples and 14 adjacent control samples from 14 patients with HCC. After cell clustering and downscaling, 112,317 cells were grouped into 25 clusters manually identified according to 10 cell types with expressed markers consistent with previous laboratory and scRNA-seq studies (Sharma et al., 2020; Sun et al., 2021; Liang et al., 2022a) (Figure 1B), including HCC cells (AMBP, TTR, ARG1, GPC3, and ALB), hepatocyte (Hep; ORM1), epithelial cells (Ep; KRT19 and MUC1), endothelial cells (En; VWF, PECAM1, ENG, CD34, CDH5, KDR, and ICAM1), fibroblasts (Fib; ACTA2, COL1A1, and PDGFRB), naïve T cells (CD3D, CD3E, and CD3G), CD8+ T cells (IFNG, CD8A, and CD8B), natural-killer cells (NK; GNLY and KLRF1), B cells (MZB1, CD79A and MS4A1), and macrophages (Mac; HLA-DRA, HLA-DRB1, HLA-DRB5, CD68, and CD14) (Figure 1C). The annotation of HCC cells was validated by inferring CNVs based on the scRNA-seq data (Figure 1D). Subsequently, by comparing the differences in cell type composition between HCC and control samples, we found naïve T cells to be more abundant in HCC than in adjacent control tissues (Figure 1E). This result suggested significant lymphatic immune infiltration of tumor cells. Additionally, the abundance of fibroblasts was higher and that of endothelial cells was lower in HCC than in adjacent control tissues. In summary, we constructed a single-cell atlas of patients with HCC, revealing differences in cell-type composition in the HCC tumor microenvironment.
FIGURE 1. Single-cell transcriptome profiling of patients with HCC. (A) Single-cell analysis process. (B) Global HCC tumor ecological map showing the different cell types. (C) Expression of marker genes for cellular annotation (size: -log10 (adjusted p-value); statistical test method: Wilcoxon rank sum). (D) The chromosomal landscape of HCC tumors. (E) The difference in composition of 10 cell types between the control and HCC groups. HCC: Hepatocellular carcinoma; TCGA-LIHC: the Cancer Genome Atlas Liver Hepatocellular Carcinoma data collection; GRN: gene regulatory network; ROC: receiver operating characteristic.
3.2 Tumor cells with drug-resistant potential are present in the HCC microenvironmentWe performed a subpopulation analysis on the HCC cells and obtained seven subpopulations, including APOA1+ HCC, SPINK1+ HCC, APOA1+SPINK1+PLA2G2A+ HCC, DDX5+ HCC, APOC2+ HCC, AIF1+ HCC, and CLDN10+ HCC (Figures 2A, B). Among these markers, APOA1 was not only highly expressed in the APOA1+ HCC subpopulation, but was also commonly expressed in other HCC subpopulations, and SPINK1 was also expressed in most HCC cells (Figure 2C). We conducted ROC curve analysis of APOA1 and SPINK1 based on the TCGA-LIHC data and found that high APOA1 and SPINK1 expression levels could serve as potential biomarkers for HCC, with AUC values of 0.782 and 0.690, respectively (Supplementary Figure S1). Enrichment analysis showed that SPINK1+ HCC and APOA1+SPINK1+PLA2G2A+ HCC cells were significantly involved in bacterial infection-related pathways (Figure 2D). Notably, all HCC subpopulations except APOC2+ HCC were significantly involved in drug metabolism-related pathways, and most of these subpopulations were at the end of the cell developmental trajectory (Figure 2E). This result suggested that as HCC progresses, tumor cells may develop drug resistance by activating drug metabolism pathways, leading to poor outcomes in patients. Therefore, targeting these subpopulations could prevent the development of drug resistance in patients. We constructed GRNs to further explore the TFs regulating the subpopulations (Figures 2F, G). The results showed that subpopulations with drug-resistant potential were mainly regulated by TFs such as CRZB5, AR, HLF, and RARA, implying that tracking these genes is helpful for understanding HCC progression. In addition, we explored the prognostic potential of specific markers highly expressed in each subpopulation of HCC cells. The results suggested that high SPINK1 and DDX5 expression levels were significantly associated with lower HCC survival Figure 2H. In summary, drug metabolism pathways may be activated by tumor cells in the HCC microenvironment, resulting in increased difficulty in treatment.
FIGURE 2. Landscape of tumor cell subpopulations in the HCC microenvironment. (A) HCC subpopulations identified in this study. (B) Markers that are specifically highly expressed in each HCC subpopulation. (C) Expression profiles of APOA1 and SPINK1 in the global single-cell landscape of HCC. (D) KEGG pathways significantly enriched in each HCC subpopulation. (E) Pseudotime differentiation trajectory of HCC subpopulations. (F) Gene regulatory network analysis of HCC subpopulations. (G) The main transcription factors regulating HCC subpopulations. (H) Kaplan-Meier plots showing worse OS prognosis in HCC with higher SPINK1 expression. (I) Kaplan-Meier plots showing worse RFS prognosis in HCC with higher DDX5 expression. HCC: hepatocellular carcinoma; OS: overall survival, RFS: relapse-free survival.
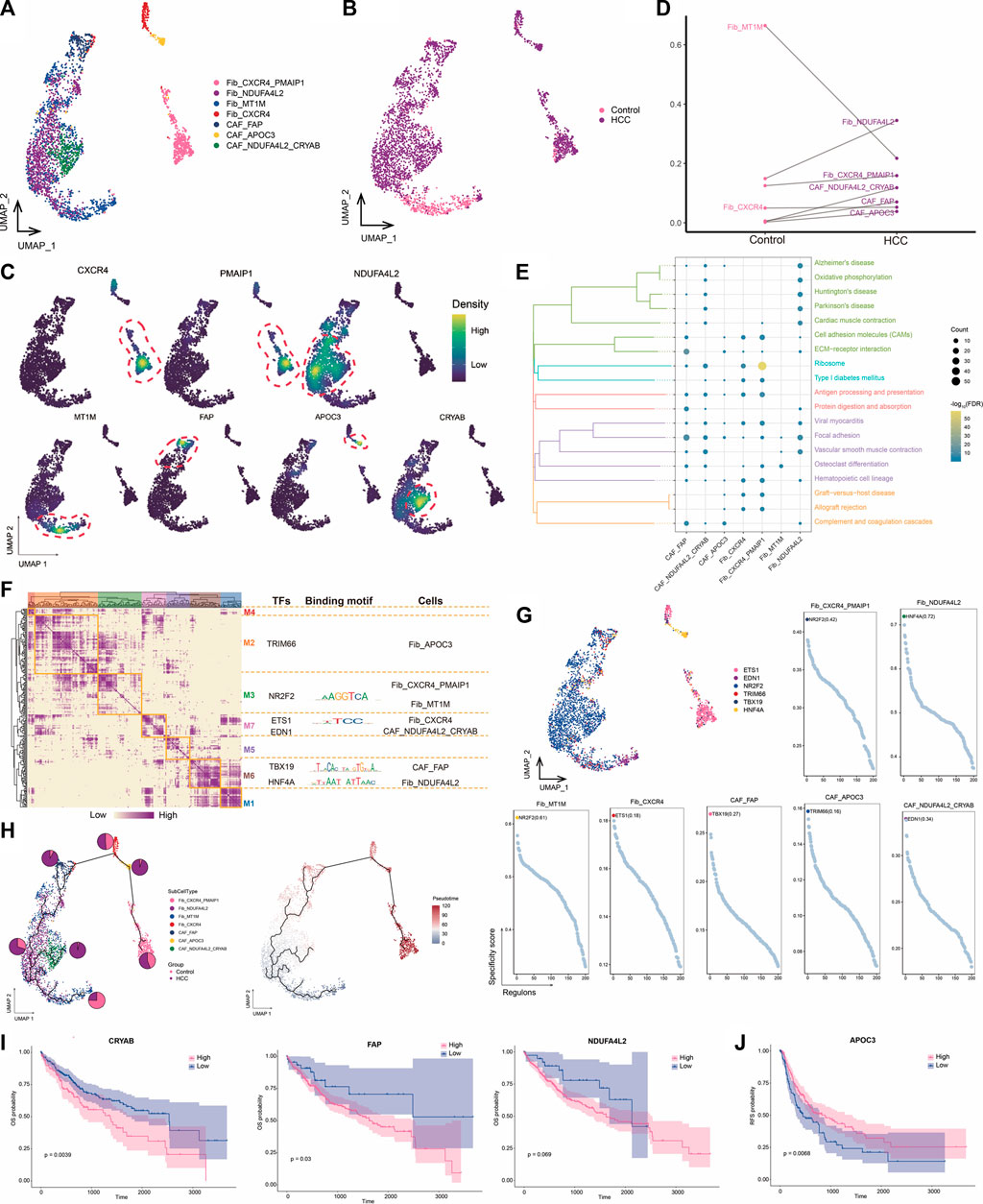
3.3 High NDUFA4L2 expression in fibroblasts may promote HCC developmentCAFs are the most abundant cell type in the TME and are the center of cross-communication between various cells in the tumor mesenchyme. Thus, we performed a subpopulation analysis and identified seven major fibroblast subpopulations expressing different markers (Figures 3A–C). We found that the abundances of NDUFA4L2+ Fib and NDUFA4L2+CRYAB+ CAF cells were significantly increased in HCC, suggesting that these subpopulations may be involved in promoting tumor development (Figure 3D). Enrichment analysis revealed that all seven subpopulations were significantly associated with the focal adhesion pathway (Figure 3E), which is the point of contact between cells and the ECM and usually closely associated with cell migration. GRN analysis of the fibroblast subpopulations revealed that the target genes were divided into seven modules regulated by TFs including NR2F2, ETS1, TBX19, and HNF4A (Figures 3F, G). Ranking of the regulator specificity scores of the fibroblast subpopulations revealed that NR2F2 was the most specific regulator of both the CXCR4+PMAIP1+ Fib and MT1M+ Fib subpopulations and has the ability to promote tumor cell proliferation, epithelial-mesenchymal transition, and invasive features (Mauri et al., 2021) (Figure 3G). As a gene therapy tool for HCC, forced re-expression of HNF4A inhibits proliferation and eliminates cancer-specific features of target cells (Takashima et al., 2018). Therefore, the combined transduction of NR2F2 and HNF4A may be more stable for cellular reprogramming in HCC.
FIGURE 3. Landscape of fibroblast subpopulations in the HCC microenvironment. (A) Fibroblast subpopulations identified in this study. (B) Cellular profiles of fibroblast subpopulations in the control and HCC groups. (C) Markers that are specifically highly expressed in each fibroblast subpopulation. (D) Differences in the abundance of fibroblast subpopulations in the control and HCC groups. (E) KEGG pathways significantly enriched in each fibroblast subpopulation. (F) Gene regulatory network analysis of fibroblast subpopulations. (G) The main transcription factors regulating fibroblast subpopulations. (H) Pseudotime differentiation trajectory of fibroblast subpopulations. (I) Kaplan-Meier plots showing worse OS prognosis in HCC with higher expression of CRYAB, FAP, and NDUFA4L2. (J) Kaplan-Meier plots showing worse RFS prognosis in HCC with lower APOC3 expression of. HCC: hepatocellular carcinoma; OS: overall survival, RFS: relapse-free survival.
Pseudotime analysis showed that NDUFA4L2+ Fib cells were positioned earlier on the differentiation trajectory than were NDUFA4L2+CRYAB+CAF cells, presumably because NDUFA4L2+ Fib cells differentiated into NDUFA4L2+CRYAB+CAFcells over time, further promoting tumor development (Figure 3H). Subsequently, we evaluated the prognostic potential of specific markers highly expressed in each subpopulation of HCC and found that patients with higher CYRAB, FAP, and NDUFA4L2 expression had poorer OS (Figure 3I), whereas patients with lower APOC3 expression had poorer RFS (Figure 3J). Therefore, high NDUFA4L2 expression in fibroblasts may promote HCC development. To address the location of fibroblasts in the TME, we need to further investigate their connections with other cells.
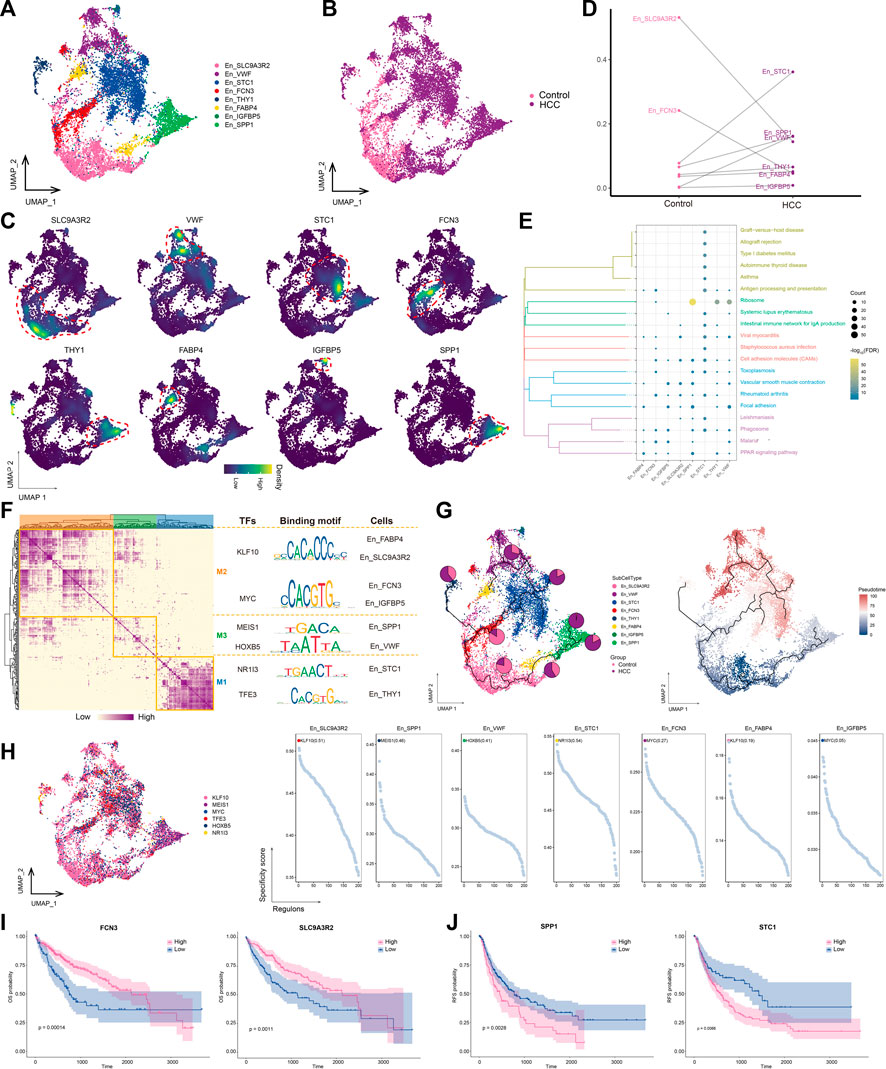
3.4 High SLC9A3R2 expression in endothelial cells differentiates into other stages of En cells as HCC developsThe transformation of endothelial cells into mesenchymal fibroblasts after stimulation, known as the endothelial-mesenchymal transition (EMT), is an important source of fibroblasts. Our analysis confirmed the increased abundance of fibroblasts and decreased abundance of endothelial cells in the HCC group (Figure 1E). Thus, we performed a subpopulation analysis of the endothelial cells and obtained eight subpopulations (Figures 4A, B), named according to the markers they specifically expressed (Figure 4C). The abundances of STC1+ En and VWF+ Encells were significantly increased in the HCC group (Figure 4D), and STC1+ En was the most abundant subtype. Most endothelial cell subpopulations were significantly associated with focal adhesion and cell adhesion molecule pathways (Figure 4E). GRN analysis of the endothelial cells revealed that the target genes were divided into three modules and regulated by TFs including KLF10, MEIS1, HOXB5, NR1I3, and TFE3 (Figure 4F). Among them, KLF10 was the most specific regulator of two subpopulations, FABP4+ En and SLC9A3R2+ En, and has important regulatory effects on perivascular fibrosis (Zhuang et al., 2022) (Figure 4G). Moreover, the SLC9A3R2+ En subpopulation, whose abundance was significantly reduced in HCC (Figure 4C), was not only significantly enriched in cell adhesion molecule pathways, but was also at an early stage of subpopulation cell differentiation, suggesting that it may gradually differentiate into other endothelial cell stages with the progression of HCC, in turn promoting tumor proliferation and migration (Figure 4H). Therefore, targeting KLF10 to regulate the abundance of the SLC9A3R2+ En subpopulation may help regulate HCC proliferation and migration. Survival analysis of marker genes showed that patients with low SLC9A3R2 and FCN3 expression had poorer OS (Figure 4I) and RFS (Figure 4J). Overall, these results suggest that SLC9A3R2+ En cells differentiate into other endothelial cell stages as HCC progresses and contribute to its further development.
FIGURE 4. Landscape of endothelial cell subpopulations in the HCC microenvironment. (A) Endothelial cell (En) subpopulations identified in this study. (B) Cellular profiles of En subpopulations in the control and HCC groups. (C) Markers that are specifically highly expressed in each En subpopulation. (D) Differences in the abundance of En subpopulations in the control and HCC groups. (E) KEGG pathways significantly enriched in each En subpopulation. (F) Gene regulatory network analysis of En subpopulations. (G) The main transcription factors regulating En subpopulations. (H) The pseudotime differentiation trajectory of En subpopulations. (I) Kaplan-Meier plots showing worse OS prognosis in HCC with lower expression of SLC9A3R2 and FCN3. (J) Kaplan-Meier plots showing worse RFS prognosis in HCC with lower expression of SLC9A3R2 and FCN3. HCC: hepatocellular carcinoma; OS: overall survival, RFS: relapse-free survival.
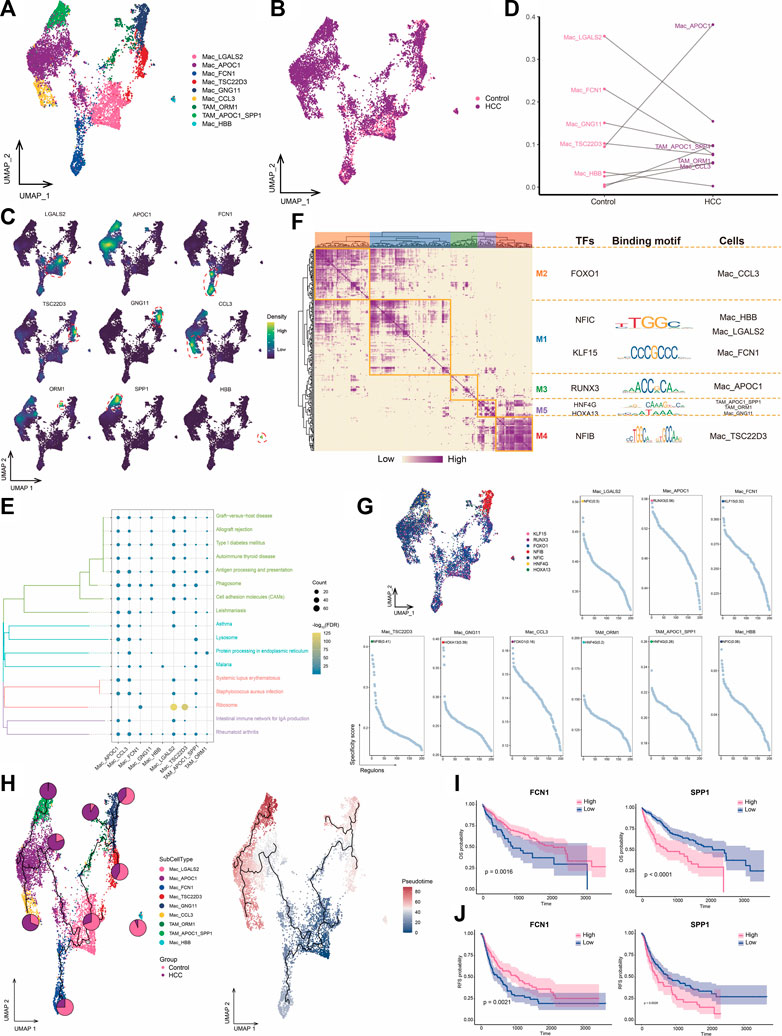
3.5 Increased infiltration of HCC tissues by macrophages highly expressing APOC1TAMs are macrophages that infiltrate tumor tissues and exert tumor-promoting and immunosuppressive effects in various ways (Wium et al., 2018; Gadiyar et al., 2020; Hedrich et al., 2021). We identified nine macrophage subpopulations and subsequently assigned names based on genes specifically highly expressed in these subpopulations (Figures 5A–C). Among them, the APOC1+ Mac subpopulation was a major component of HCC macrophage ecology compared to levels in adjacent control tissue, whereas the abundance of the FCN1+ Mac subpopulation was significantly decreased in tumor cells. The APOC1+SPP1+ TAM and HSPA1B+ TAM subpopulations were particularly abundant in the HCC group and were therefore defined as TAMs (Figure 5D). Most macrophage subpopulations were significantly involved in pathways related to antigen processing and expression, suggesting an active immune function of macrophages in the HCC microenvironment (Figure 5E). GRN analysis of macrophages revealed that the target genes were divided into five modules regulated by TFs including NFIC, KLF15, RUNX3, HNF4G, HOXA13, and NFIB (Figures 5F, G). Pseudotime analysis showed similar differentiation trajectories for APOC1+ Mac and APOC1+SPP1+ TAM cells, which were significantly more abundant in HCC, suggesting increased infiltration by macrophages highly expressing APOC1 with the development of HCC (Figure 5H). Macrophage subpopulations with high APOC1 expression were found at the end of the cell developmental trajectory and specifically expressed the M2 macrophage marker CD163 (Supplementary Figure S2), suggesting that these subpopulations may accelerate HCC development. Survival analysis of the subpopulation-specific markers showed that FCN1 and SPP1 had prognostic potential for HCC (Figures 5I, J). In HCC, the presence of TAM subpopulations correlates with poor outcomes (Graham and Pollard, 2022). In our study, TAM subpopulations were positioned at the end of the cell developmental trajectory and were regulated by the same TFs, implying that macrophage cell states could be changed to prevent the development of tumor cells.
FIGURE 5. Landscape of macrophage subpopulations in the HCC microenvironment. (A) Macrophage subpopulations identified in this study. (B) Cellular profiles of macrophage subpopulations in the control and HCC groups. (C) Markers that are specifically highly expressed in each macrophage subpopulation. (D) Differences in the abundance of macrophage subpopulations in the control and HCC groups. (E) KEGG pathways significantly enriched in each macrophage subpopulation. (F) Gene regulatory network analysis of macrophage subpopulations. (G) The main transcription factors regulating macrophage subpopulations. (H) Pseudotime differentiation trajectory of macrophage subpopulations. (I) Kaplan-Meier plots showing the relationship between OS prognosis in HCC and FCN1 and SPP1 expression. (J) Kaplan-Meier plots showing the relationship between RFS prognosis in HCC and FCN1 and SPP1 expression. HCC: hepatocellular carcinoma; OS: overall survival, RFS: relapse-free survival.
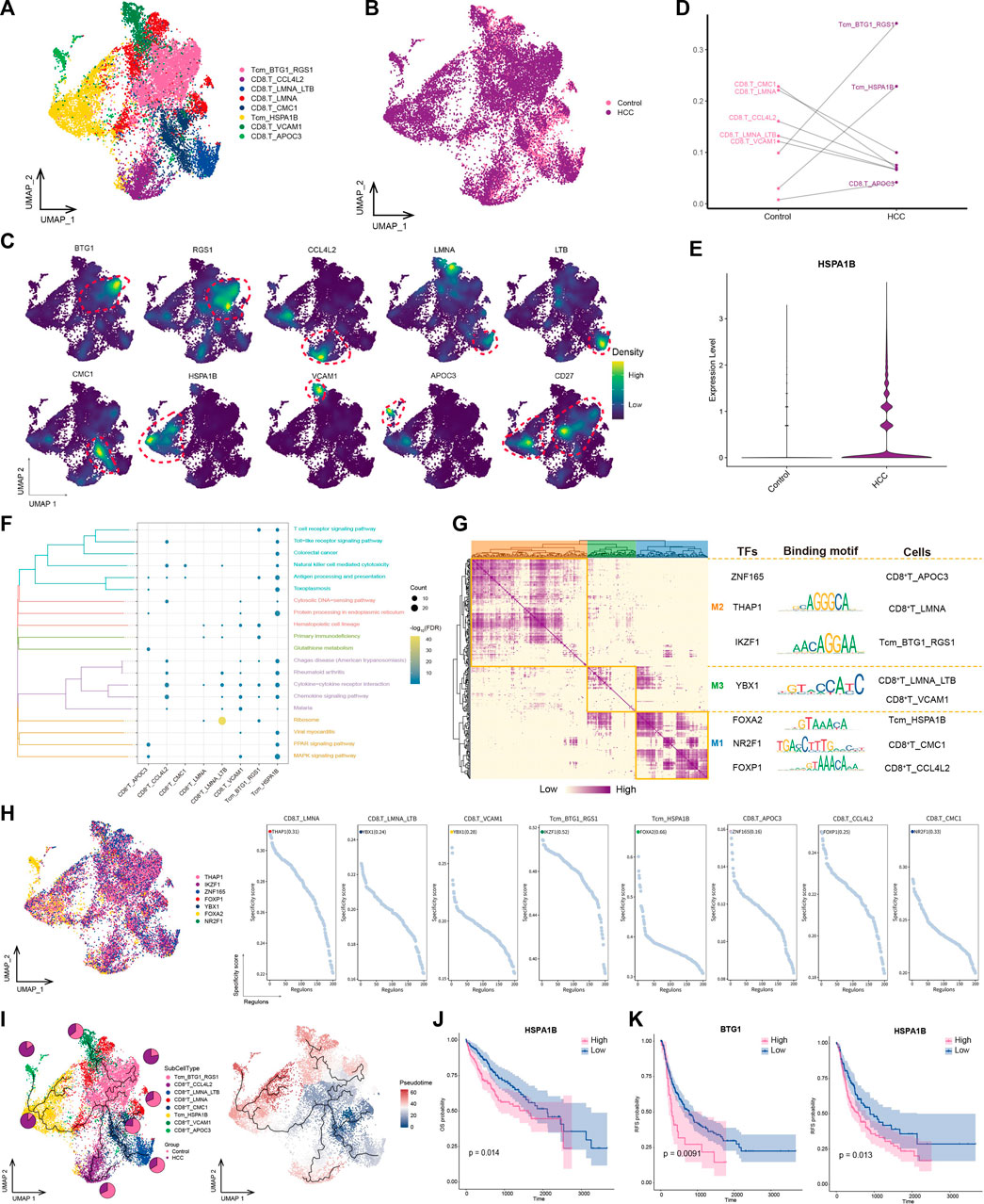
3.6 Central memory T cells with high HSPA1B expression play a significant role in HCC immunityT lymphocytes play an important effector-killing role in antitumor immunity. To explore the differences between lymphocytes in tumors and normal tissues, we identified eight CD8+ T cell subpopulations (Figures 6A, B) expressing different specific markers (Figure 6C). Among them, CD27 was specifically expressed in the BTG1+RGS1+ Tcm and HSPA1B+ Tcm subpopulations, thus classifying them as subpopulations of central memory T cells (Tcms) (Figure 6E). The subpopulations with double-positive expression of BTG1 and RGS1 were significantly enriched in tumor lesions (Figure 6D). In addition, the HSPA1B+ Tcm subpopulation was significantly enriched in the HCC group, and HSPA1B was significantly enriched in all cell types in the HCC group (Figure 6C), suggesting its association with the remodeling of the tumor ecological niche in HCC. Enrichment analysis showed that most CD8+ T cell subpopulations were significantly involved in chemokine-and natural killer-mediated cytotoxicity-related pathways, especially HSPA1B+ Tcm, which was significantly enriched in HCC. This result suggested that the subpopulation may be immunologically active in the HCC microenvironment, aggregating to lesions and exerting immunocidal effects (Figure 6F).
FIGURE 6. Landscape of CD8+ T subpopulations in the HCC microenvironment. (A) CD8+ T subpopulations identified in this study. (B) Cellular profiles of CD8+ T subpopulations in the control and HCC groups. (C) Markers that are specifically highly expressed in each CD8+ T subpopulation. (D) Differences in the abundance of CD8+ T subpopulations in the control and HCC groups. (E) Expression of HSPA1B in the control and HCC groups. (F) KEGG pathways significantly enriched in each CD8+ T subpopulation. (G) Gene regulatory network analysis of CD8+ T subpopulations. (H) The main transcription factors regulating CD8+ T subpopulations. (I) Pseudotime differentiation trajectory of CD8+ T subpopulations. (J) Kaplan-Meier plots showing worse OS prognosis in HCC with higher HSPA1B expression. (K) Kaplan-Meier plots showing worse RFS prognosis in HCC with higher HSPA1B and BTG1 expression. HCC: hepatocellular carcinoma; OS: overall survival, RFS: relapse-free survival.
GRN analysis of CD8+ T cells showed that the target genes were divided into three modules regulated by TFs including THAP1, IKZF1, YBX1, FOXA2, NR2F1, and FOXP1 (Figures 6G, H). Pseudotime analysis showed that HSPA1B+ Tcm was positioned at a late stage of development, suggesting that as HCC develops, this subpopulation of cells is stimulated by antigens, proliferates, and differentiates to exert antitumor effects (Figure 6I). Survival analysis revealed that among the subpopulation-specific markers, BTG1 and HSPA1B had prognostic potential for HCC (Figures 6J, K). In conclusion, HSPA1B+ Tcm subpopulation plays an important role in the immune response to HCC, suggesting that increasing the abundance of this subpopulation may help inhibit tumor growth and metastasis.
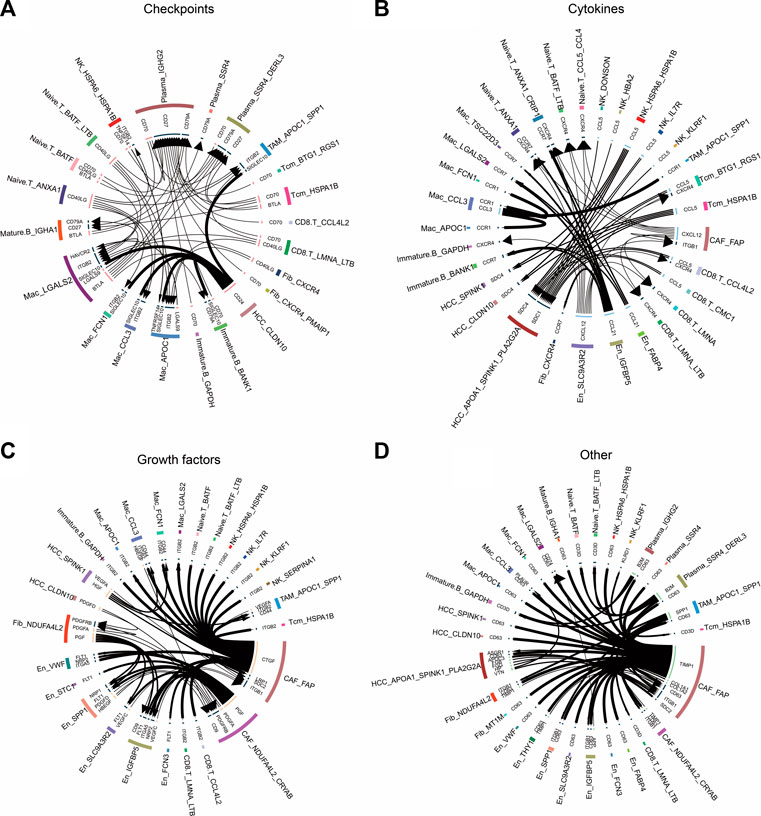
3.7 Cellular communication events in the HCC microenvironmentTo explore the relationship between non-tumor and tumor cells, we inferred their receptor–ligand interactions using the iTALK approach and constructed a cellular communication network divided into four modules: cytokines, growth factors, immune checkpoints, and other (Figure 7). We found that the CCL5 ligand secreted by BTG1+RGS1+ Tcm and HSPA1B+ Tcm cells bound to the SDC4/1 receptor on the surfaces of tumor cells (Figure 7B). Interestingly, the checkpoint CD24 ligand secreted by tumor cells bound to the SIGLEC10 receptor on macrophages to transmit inhibitory signals and reduce phagocytosis (Figure 7A). We also observed that tumor and endothelial cells released vascular endothelial growth factors, including VEGFC and VEGFA, as well as platelet-derived growth factors, including PDGFA and PDGFC (Figure 7C). Remarkably, in another module, the TIMP1 ligand secreted by CAFs bound to the receptor CD63 on the surfaces of tumor cells and TAMs, whereas the SPP1 ligand secreted by APOC1+SPP1+ TAM cells bound to the ITGB1 receptor on CAFs (Figure 7D). These cellular communication networks reshape the tumor microenvironment and promote the development of HCC.
FIGURE 7. Circle plots demonstrate the ligand–receptor interactions between cells. (A) Receptor–ligand pairs related to immune checkpoints for each cell type. (B) Receptor–ligand pairs related to cytokines for each cell type. (C) Receptor–ligand pairs related to growth factors for each cell type. (D). Other receptor–ligand pairs for each cell type, excluding immune checkpoints, cytokines, and growth factors.
4 DiscussionIn this study, we constructed a single-cell microenvironmental landscape of patients with primary HCC based on scRNA-seq data to reveal cell subpopulations with potentially specific functions in the HCC tumor microenvironment and to explore the interactions between HCC and the tumor microenvironment. High levels of CNVs are present in HCC tissue samples and have been suggested as potential factors in HCC development. Additionally, the significant increase in specific cell subpopulations compared to levels in controls may be related to HCC formation.
Previous studies have shown that elevated levels of APOA1 are associated with liver injury (Gumilas et al., 2022). In the present study, APOA1 was not only most highly expressed in the APOA1+ HCC subpopulation, but was also highly expressed in other malignant HCC cells; therefore, it may be a potential biomarker for HCC. Pseudotime analysis suggested the APOA1+ HCC subpopulation to be the origin of tumor development, which again demonstrates the potential of APOA1 as a marker for early HCC detection. We found that HIF, a TF that induces hypoxic gene expression and helps tumor cells adapt to hypoxic environments, was highly expressed in the APOA1+ HCC subpopulation (Balamurugan, 2016). In addition, we found that hypoxia-related genes, such as ALDOB, GPC3, CP, PRDX5, and FBP1, were highly expressed in the DDX5+ HCC subpopulation. Hypoxia stimulates development of the EMT, and the CAF subpopulation in the present study also highly expressed DDX5. DDX5 is a member of the DEAD-BOX family of RNA-unwinding enzymes, and its knockdown can promote migration, invasion, and EMT processes in HCC cells (Xue et al., 2018). Thus, these findings complement the potential mechanism underlying the oncogenic effects of DDX5.
Cell adhesion to the ECM is a key determinant among the myriad microenvironmental factors that influence drug resistance in cancer cells (Eke and Cordes, 2015). In the present study, we found that all fibroblast subpopulations were significantly involved in cell motility-related pathways, including the focal adhesion pathway. The abundance of the NDUFA4L2+ Fib subpopulation was significantly higher in HCC. NDUFA4L2 expression is positively correlated with tumor stage, and patients with higher expression have worse RFS than those with lower expression (Sarathi and Palaniappan, 2019). Pseudotime analysis showed that NDUFA4L2+ Fib cells were positioned earlier on the differentiation trajectory than were NDUFA4L2+CRYAB+ CAF cells, presumably because NDUFA4L2+ Fib cells gradually differentiated into NDUFA4L2+CRYAB+ CAF cells with the development of HCC. In addition, we found that FAP+ CAF bound to CD63 in tumor cells by secreting the TIMP1 ligand. TIMP1 is a multifunctional protein that promotes cell proliferation, growth, survival, and differentiation and suppresses cell apoptosis in a variety of different tumor types (Justo and Jasiulionis, 2021); thus, the TIMP1–CD63 interaction between CAFs and HCC cells may promote the proliferation of HCC cells. Previous studies have shown that FAP+ CAFs and SPP1+ TAMs contribute to ECM remodeling and coordinate the formation of a prodesmoplastic microenvironment that prevents lymphocytes from infiltrating the tumor core (Qi et al., 2022). In our study, we found that SPP1 secreted by APOC1+SPP1+ TAM cells bound to ITGF1 secreted by FAP+ CAFs cells, suggesting that these two subpopulations may promote HCC development by remodeling the tumor microenvironment. Additionally, the higher abundance of naïve T cells in the HCC group than in the control group implied significant lymphoid immune infiltration of HCC tissues. FAP+ CAF interacted with naïve T cells via the CXCL12–CXCR4 axis. CXCL12 is a homeostatic chemokine that regulates physiological and pathological processes such as inflammation, cell proliferation, and specific migration. TME enriched with CXCL12 is resistant to immune checkpoint inhibitor treatment (Portella et al., 2021). In this study, CAFs were the source of CXCL12 signaling, suggesting the importance of CAF research in the treatment of HCC with immune checkpoint inhibitors.
The STC1+ En subpopulation was significantly enriched in the HCC group, with the highest abundance among the subpopulations identified in this study. Moreover, we found that high STC1 expression is significantly associated with poor HCC prognosis. VWF promotes chronic hepatitis B virus- and hepatitis C virus-related HCC (Xiang et al., 2022). Another study showed that HCC is associated with significant changes in primary hemostasis, including increased platelet aggregation and elevated VWF levels (Zanetto et al., 2021). VWF participates in angiogenesis and negative regulation of angiogenic factors, which is an essential link in the growth, invasion, and metastasis of HCC. In the present study, the VWF+ En subpopulation was significantly increased in the HCC group, suggesting that VWF may play a role in promoting angiogenesis in HCC.
The APOC1+ Mac subpopulation was a major component of the macrophage ecology of HCC and was significantly more abundant compared to the levels in adjacent controls. APOC1 expression was also higher in the APOC1+SPP1+ TAM subpopulation, and this gene was specifically enriched in HCC lesions. It has been reported that APOC1 is overexpressed in the TAMs of HCC tissues compared to those in normal tissues, and inhibition of APOC1 promotes the transformation of M2 macrophages into M1 macrophages through the iron death pathway to reshape the tumor immune microenvironment (Hao et al., 2022), indicating the pro-cancer role of APOC1 in HCC. Our study showed that APOC1 was not only highly expressed in macrophages in HCC lesions, but also that macrophages with high APOC1 expression were more abundant in HCC lesions than were other macrophages. Additionally, patients with high SPP1 expression have poor prognosis, and SPP1 is a pro-cancer gene in HCC (Liu et al., 2022). We found that SPP1 was specifically expressed in TAM subpopulations, suggesting that it may function through macrophages. The FCN1+ Mac subpopulation was significantly less abundant in tumor cells than in adjacent controls. FCN1 is a member of the ficolin (FCN) family of proteins and a part of the innate immune system (Zhang et al., 2010). The results of the present study were consistent: lower FCN1 expression was associated with a lower patient survival rate (Sun et al., 2022).
The HSPA1B+ Tcm subpopulation was significantly increased in the HCC group, and HSPA1B was significantly enriched in all cell types in the HCC group, suggesting its involvement in remodeling of the tumor ecological niche in HCC. HSPA1B belongs to heat shock protein family A, where the binding of other heat shock proteins can stabilize existing proteins and mediate the folding of newly translated proteins in the cytosol and organelles (Faisal et al., 2022). Moreover, HSPA1B is involved in the ubiquitin-proteasome pathway through interaction with the AU-rich element RNA-binding protein 1, and the ubiquitin-proteasome system is an important non-lysosomal protein degradation pathway in cells that directly or indirectly affects the occurrence of various malignant tumors by regulating cell cycle activity and apoptosis-related proteins and activating or inhibiting the expression of proto-oncogenes and anti-oncogenes (Park et al., 2020). In addition, previous studies have shown that the receptor CCL5–SDC1/4 ligand interactions between regulatory T cells and tumor cells in pancreatic cancer promote tumor cell metastasis (Chen et al., 2022). The same intercellular communication signals were also observed in this study, where the CCL5 ligand secreted by BTG1+RGS1+ Tcm bound to SDC4/1 receptors on the surfaces of tumor cells, suggesting that the interaction between BTG1+RGS1+ Tcms and tumor cells via CCL5–SDC4/1 axis may promote tumor progression. These findings may provide new targets for HCC immunotherapy.
This study preliminarily revealed the cell subsets that lead to HCC in the tumor microenvironment based on scRNA-seq data; however, some limitations remain. First, only a single dataset was included in this study, and the sample size was relatively small. In addition, the conclusions of this study were mainly based on bioinformatics analysis; therefore, experimental validation with larger sample sizes is warranted.
In conclusion, our study suggests the presence of tumor cells with drug-resistant potential in the HCC microenvironment. Among non-tumor cells, high NDUFA4L2 expression in fibroblasts may promote tumor progression, whereas high HSPA1B expression in Tcms may exert antitumor effects. In addition, there is significant immune infiltration into tumor lesions, and the interaction between BTG1+RGS1+ Tcms and tumor cells via CCL5–SDC4/1 axis may promote tumor progression. Focusing on the role of CAFs and TAMs, which are closely related to tumor cells, in tumors would be beneficial to the progress of systemic therapy research.
Data availability statementThe original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributionsAll authors contributed to the study conception and design. Bioinformatics analysis were performed by JG and ZL. The first draft of the manuscript was written by JG, and all authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
FundingThis work was supported by the Guangxi Science and Technology Base and Talent Special Fund (AB23026002, AD22035042), National Natural Science Foundation of China (82060512, 31471271, 31560311), Guangxi Natural Science Fund for Innovation Research Team (2016GXNSFGA380006), Guangxi science and technology development project (AD17195090, AB18126055), Natural Science Foundation of Guangxi Province of China (2020GXNSFAA259022), Innovation Project of Guangxi Graduate Education (YCSW2021139), International Communication of Guangxi Medical University Graduate Education.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1194199/full#supplementary-material
ReferencesBecht, E., Mcinnes, L., Healy, J., Dutertre, C. A., Kwok, I. W. H., Ng, L. G., et al. (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44. doi:10.1038/nbt.4314
PubMed Abstract | CrossRef Full Text | Google Scholar
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. doi:10.1038/nbt.4096
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, K., Wang, Y., Hou, Y., Wang, Q., Long, D., Liu, X., et al. (2022). Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and tumor cells in pancreatic cancer. Cancer Lett. 545, 215834. doi:10.1016/j.canlet.2022.215834
PubMed Abstract | CrossRef Full Text | Google Scholar
D'Souza, S., Lau, K. C., Coffin, C. S., and Patel, T. R. (2020). Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterology 26, 5759–5783. doi:10.3748/wjg.v26.i38.5759
CrossRef Full Text | Google Scholar
Faisal, S., Abdelaal, S., Jeraiby, M. A., Toaimah, F. H. S., Kattan, S. W., Abdel-Gawad, A. R., et al. (2022). Diagnostic and prognostic risk assessment of heat shock protein HSPA1B rs2763979 gene variant in asthma. Genes 13, 2391. doi:10.3390/genes13122391
PubMed Abstract | CrossRef Full Text | Google Scholar
Gadiyar, V., Patel, G., and Davra, V. (2020). Immunological role of TAM receptors in the cancer microenvironment. Int. Rev. Cell Mol. Biol. 357, 57–79. doi:10.1016/bs.ircmb.2020.09.011
PubMed Abstract | CrossRef Full Text | Google Scholar
Graham, N., and Pollard, J. W. (2022). An acid trip activates protumoral macrophages to promote hepatocellular carcinoma malignancy. J. Clin. Invest 132, e158562. doi:10.1172/JCI158562
PubMed Abstract | CrossRef Full Text | Google Scholar
Gumilas, N. S. A., Harini, I. M., Ernawati, D. A., Indriani, V., Novrial, D., and Kurniawan, D. W. (2022). Potential of apolipoprotein A1 (ApoA1) for detecting liver cirrhosis and hepatocellular carcinoma. Asian Pac. J. cancer Prev. APJCP 23, 2001–2008. doi:10.31557/APJCP.2022.23.6.2001
CrossRef Full Text | Google Scholar
Hafemeister, C., and Satija, R. (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296. doi:10.1186/s13059-019-1874-1
PubMed Abstract | CrossRef Full Text | Google Scholar
Hao, X., Zheng, Z., Liu, H., Zhang, Y., Kang, J., Kong, X., et al. (2022). Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 56, 102463. doi:10.1016/j.redox.2022.102463
PubMed Abstract | CrossRef Full Text | Google Scholar
Hedrich, V., Breitenecker, K., Djerlek, L., Ortmayr, G., and Mikulits, W. (2021). Intrinsic and extrinsic control of hepatocellular carcinoma by TAM receptors. Cancers (Basel) 13, 5448. doi:10.3390/cancers13215448
PubMed Abstract | CrossRef Full Text | Google Scholar
Justo, B. L., and Jasiulionis, M. G. (2021). Characteristics of TIMP1, CD63, and β1-integrin and the functional impact of their interaction in cancer. Int. J. Mol. Sci. 22, 9319. doi:10.3390/ijms22179319
PubMed Abstract | CrossRef Full Text | Google Scholar
Kumar, V., Xin, X., Ma, J., Tan, C., Osna, N., and Mahato, R. I. (2021). Therapeutic targets, novel drugs, and delivery systems for diabetes associated NAFLD and liver fibrosis. Adv. Drug Deliv. Rev. 176, 113888. doi:10.1016/j.addr.2021.113888
PubMed Abstract | CrossRef Full Text | Google Scholar
Liang, J., Chen, W., Ye, J., Ni, C., and Zhai, W. (2022a). Single-cell transcriptomics analysis reveals intratumoral heterogeneity and identifies a gene signature associated with prognosis of hepatocellular carcinoma. Biosci. Rep. 42. doi:10.1042/BSR20212560
CrossRef Full Text | Google Scholar
Liang, Y., He, H., Wang, W., Wang, H., Mo, S., Fu, R., et al. (2022b). Malignant clonal evolution drives multiple myeloma cellular ecological diversity and microenvironment reprogramming. Mol. Cancer 21, 182. doi:10.1186/s12943-022-01648-z
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, L., Zhang, R., Deng, J., Dai, X., Zhu, X., Fu, Q., et al. (2022). Construction of TME and Identification of crosstalk between malignant cells and macrophages by SPP1 in hepatocellular carcinoma. Cancer Immunol. Immunother. CII 71, 121–136. doi:10.1007/s00262-021-02967-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Mauri, F., Schepkens, C., Lapouge, G., Drogat, B., Song, Y., Pastushenko, I., et al. (2021). NR2F2 controls malignant squamous cell carcinoma state by promoting stemness and invasion and repressing differentiation. Nat. Cancer 2, 1152–1169. doi:10.1038/s43018-021-00287-5
PubMed Abstract | CrossRef Full Text | Google Scholar
Park, J., Cho, J., and Song, E. J. (2020). Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Archives Pharmacal Res. 43, 1144–1161. doi:10.1007/s12272-020-01281-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Qi, J., Sun, H., Zhang, Y., Wang, Z., Xun, Z., Li, Z., et al. (2022). Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 13, 1742. doi:10.1038/s41467-022-29366-6
PubMed Abstract | CrossRef Full Text | Google Scholar
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi:10.1093/nar/gkv007
PubMed Abstract | CrossRef Full Text | Google Scholar
Robin, X., Turck, N., Hainard, A., Tiberti, N., Lisacek, F., Sanchez, J. C., et al. (2011). pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinforma. 12, 77. doi:10.1186/1471-2105-12-77
PubMed Abstract | CrossRef Full Text | Google Scholar
Sarathi, A., and Palaniappan, A. (2019). Novel significant stage-specific differentially expressed genes in hepatocellular carcinoma. BMC cancer 19, 663. doi:10.1186/s12885-019-5838-3
PubMed Abstract | CrossRef Full Text | Google Scholar
Sharma, A., Seow, J. J. W., Dutertre, C.-A., Pai, R., BléRIOT, C., Mishra, A., et al. (2020). Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 183, 377–394. doi:10.1016/j.cell.2020.08.040
留言 (0)