
記住我





Vascular formation is regulated by various growth factors, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and transforming growth factor-β (TGFβ), which signal to endothelial cells (ECs) through their receptor kinases (Gerhardt et al., 2003; Shih et al., 2003; Ma et al., 2020; Chen et al., 2021). Dysregulation of these signaling pathways may lead to developmental errors during embryogenesis and can cause vascular anomalies, such as capillary malformations or arteriovenous malformations (AVM) (Hellstrom et al., 1999; Cunha et al., 2017; Chen et al., 2021). After the development of the primitive vascular plexus (Goldie et al., 2008), the ECs begin to differentiate into arterial or venous ECs (arterial-venous specification) (Swift and Weinstein, 2009; Kume, 2010). VEGF and Notch signaling play an important role for the regulation of the artery-specific gene expression, whereas COUP-TFII regulates venous-specific gene expression through the inhibition of Notch activity (Krebs et al., 2000; Fischer et al., 2004; You et al., 2005; Gridley, 2007; Swift and Weinstein, 2009). The newly formed arterial ECs then recruit vascular smooth muscle cells (vSMCs) to stabilize the vessels (Gaengel et al., 2009; Stratman et al., 2020). PDGF and angiopoietins are known regulators of vSMC recruitment during vascular development and remodeling (Hellstrom et al., 1999; Gaengel et al., 2009). Mice with genetic loss of Pdgfb or its receptor, Pdgfrb, die perinatally with vSMC recruitment defects (Levéen et al., 1994; Lindahl et al., 1997). Mice lacking ANGIOPOIETIN1 (encoded by Angpt1) or its receptor TIE2 (encoded by Tek) show lethality at an early embryonic stage and show a lack of vSMC recruitment, indicating that the ANGIOPOIETIN1-TIE2 signaling pathway plays an important role for vSMC recruitment (Dumont et al., 1994; Sato et al., 1995; Suri et al., 1996). In contrast to the TIE2 activating functions of ANGIOPOIETIN1, ANGIOPOIETIN2 (encoded by Angpt2) is known as a functional antagonist of ANGIOPOIETIN1-TIE2 signaling pathway because its overexpression phenocopies Angpt1 or Tek KO mice (Maisonpierre et al., 1997; Gale et al., 2002). However, the molecular mechanisms controlling the expression of these genes remain unclear.
PRDM16 is a member of the PRDM family of transcription factors that are defined by the presence of an active PRDI-BF1-RIZ1 methyltransferase domain (Fog et al., 2012; Pinheiro et al., 2012). PRDM16 has been studied for its role in the regulation of cell fate between muscle and brown fat cells (Seale et al., 2007; Borensztein et al., 2012; Becerril et al., 2013; Cohen et al., 2014; Harms et al., 2014; Li et al., 2015; Gan et al., 2018; Biferali et al., 2021). Also, PRDM16 has roles in craniofacial and cardiac development as well as hematopoietic stem cell maintenance (Bjork et al., 2010; Chuikov et al., 2010; Aguilo et al., 2011; Birjiniuk et al., 2018; Bjork et al., 2018; Corrigan et al., 2018; Cibi et al., 2020; Nam et al., 2020; Shull et al., 2020). Global Prdm16 knockout mice display respiratory failure and perinatal death (Bjork et al., 2010). In ECs, PRDM16 is highly expressed in the arterial ECs, and is mediated by LMO2 that associates to the Prdm16 promoter region in zebrafish (Matrone et al., 2021). In mice, PRDM16 is indispensable for the recovery of arterial flow upon femoral artery ligation due to the role of PRDM16 for maintaining endothelial function (Craps et al., 2021). A recent study has shown that PRDM16 is expressed in the retinal arteries of adult mice (Craps et al., 2021); however, a detailed analysis of PRDM16 during development and angiogenesis remains unclear.
Here, we investigate the expression of PRDM16 in developing mouse embryos and retinas. We found that loss of Prdm16 in ECs induces ectopic venous marker expression and proliferation in the arterial ECs and less vSMC recruitment around the arterial vessels. We also found that the expression of Angpt2 is upregulated in EC-specific Prdm16 knockout mice, suggesting that PRDM16 may have a crucial role for proper arterial vascular development.
Materials and methodsAnimalsAll animal experiments were performed with the approval of the institutional animal care and use committee of Cincinnati Children’s Hospital Medical Center. Mouse lines harboring the Prdm16 floxed alleles, PDGFb-ERT2-Cre (PDGFbCre), and Tek/Tie2-Cre (Tie2Cre) have been previously described (Kisanuki et al., 2001; Claxton et al., 2008; Cohen et al., 2014). Embryos were collected from pregnant Prdm16flox/flox females at the predicted embryonic time point after breeding with a Prdm16flox/+; Tie2-Cre male mouse. These alleles were kept on mixed backgrounds. Tamoxifen (Sigma) was dissolved in 90% sunflower oil (Sigma) and 10% ethanol to 2 mg/mL. Cre induction by tamoxifen was performed via intraperitoneal (IP) injection from postnatal (P) 1-3, with 0.1 mg tamoxifen injected on each day.
Whole-mount immunostaining of retinasRetinas were collected as previously described (Crist et al., 2017). Briefly, neonates were sacrificed by isoflurane overexposure and eyes were removed and fixed with 4% paraformaldehyde (PFA, Thermo Scientific) in phosphate buffered saline (PBS, Fisher Scientific) for 1h at 4°C, then washed with PBS. Retinas were dissected and washed with PBS, permeabilized with 1% Triton-X100 (Fisher Scientific) in PBS for 30 min at RT, then blocked with CAS-Block (Life Technologies) for 30 min at RT. Retinas were incubated with primary antibodies (Supplementary Table S1) or IB4 conjugated with Alexa Fluor 488 (ThermoFisher, 1:100) overnight in 1% Triton-X100 in PBS at 4°C on a rocker. Retinas were washed with PBS, then incubated with secondary antibodies (Supplementary Table S1) for 4 hs at RT on a nutator mixer. Retinas were washed with PBS, then leaflets were cut into the retinas for flat mounting on slides under a coverslip with Fluoromount-G (SouthernBiotech). Images were taken using Eclipse Ti Confocal microscopy with a C2 laser-scanning head (Nikon). Images showing the superficial, intermediate, and deep layers were stacked using ImageJ (Schneider et al., 2012). Depth coded images were prepared using the “Temporal-Color Coder”, provided by ImageJ. ENDOMUCIN and COUP-TFII mean fluorescence images were quantified using ImageJ.
EdU incorporation5 μg/kg 5-ethynyl-2′-deoxyuridin (EdU, Invitrogen) was injected into mice at P7 via IP injection. 24 hs later, retinas were collected, and then fixed with 4% PFA, blocked with CAS-Block, and incubated with anti-ERG antibody. EdU was then labeled using the Click-iT EdU 488 imaging kit (Invitrogen). Retinas were then washed in PBS at RT on a shaker, then proceeded to secondary antibody treatment, as previously detailed. To quantify the proliferating ECs, we counted EdU and ERG double positive cells on the main vessel trunk between the optic nerve and 50% vascular extension.
Isolation of brain ECsP5 mice were knocked down using ice and decapitated. Brains were removed and placed in ice cold PBS. Enzymes from the Neural Tissue Dissociation Kit (P) (MACS Miltenyi Biotec) were added to the brains and were then digested using the gentleMACS Octo Dissociator with Heaters following the manufacturer’s protocol. The dissociated cell suspension was then incubated with beads conjugated with anti-CD31 antibody (MACS Miltenyi Biotec) and applied to a magnetic column (MACS Miltenyi Biotec). CD31+ cells were eluted from the column for RNA isolation.
RNA isolation and RT-qPCRRNA from CD31+ cells were isolated according to the NucleoSpin RNA XS kit (Macherey-Nagel), then stored at −80°C until analysis. RNA from P9 retinas were isolated using TRIzol (Invitrogen) following the manufacture’s protocol. RNA concentration was assessed by NanoDrop (ThermoFisher). cDNA was generated using PrimeScript RT MasterMix (Takara). Briefly, 50 ng or 500 ng of total RNA for CD31+ cells or retinas, respectively, were used according to the manufacturer’s protocol. qPCR was performed with PowerUp SYBR Green Master Mix (applied biosystems) with primers as detailed in Supplementary Table S2. Data was collected on a Step One Plus RT-PCR system (applied biosystems). Pecam1 served as the internal control for the qPCR analysis of retina and isolated brain EC, while 18s was used for the internal control for the qPCR analysis of HUVECs.
RNA sequencing and data processingPolyA RNA-seq was performed as previously described (Walsh et al., 2019; Rapp et al., 2020). Briefly, total RNA quality was checked by Bioanalyzer (Agilent). RNA was prepared from 100 ng total RNA with NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB), then was enriched with SMARTer Apollo NGS library system (Takara), and the library was prepared using NEBNext Ultra II Directional RNA library prep kit (NEB) with 13 PCR cycles. Libraries were checked for quality and quantified with Qubit (ThermoFisher), then pooled and sequenced with NextSeq 550 sequencer (Illumina) to generate 25 M reads per sample. Fastq files were generated with Illumina BaseSpace Sequence Hub. Differentially expressed genes were identified via BaseSpace RNA-Seq Alignment v2.02, followed by RNA-Seq Differential Expression app v 1.0.1. Reads were aligned to Mus. Musculus/MM10 under 1st strand setting, analyzed using STAR for alignment and Salmon for quantification. Alignment results were analyzed in RNA-Seq Differential Expression app. RNA sequencing was performed by Genomics, Epigenomics and Sequencing Core, University of Cincinnati.
Cell cultureHuman Umbilical Vein Endothelial Cells (HUVECs) were cultured at 37°C, 5% CO2 in EGM-2 medium (Lonza).
Lentivirus generation and infectionPrdm16 was subcloned from pcDNA3.1-Prdm16, a gift from Bruce Spiegelman (Addgene plasmid # 15503 (Seale et al., 2007)), into pLVX lentiviral vector. Lentivirus for overexpression of Prdm16 was generated as previously described (Sakabe et al., 2017). Briefly, pLVX with or without Prdm16 was co-transfected into HEK293-FT cells, along with psPAX2 (packaging vector) and pMD2. G (envelope vector), while cells were cultured in DMEM (Invitrogen) with 10% FBS (R&D). Viral supernatants were collected 24 and 48 h post-transfection, pooled together and subjected to ultracentrifugation at 25,000 RPM for 2 h at 4°C. Supernatant was carefully removed and the viral pellet was suspended in 100 µL residual DMEM with 10% FBS overnight at 4°C. HUVECs were infected in a 6-well plate (Falcon) with 10 µL of control or Prdm16 virus in EGM-2 supplemented to 8 μg/mL Polybrene for 24 h. Infected HUVECs were maintained with EGM-2 and lysed with 2x sample buffer (Bio-Rad) with β-mercaptoethanol 4 days post-infection.
Western blottingInfected HUVECs were lysed with 200 µL 2x sample buffer with β-mercaptoethanol and boiled at 95°C for 5 min. 10µL of protein sample was loaded to a 6% or 10% SDS-PAGE gel, followed by transfer to Immobilon-P PVDF membrane (Millipore). Membranes were briefly washed and then blocked with 5% skim milk in wash buffer for 30 min. Membranes were incubated overnight at 4°C with anti-PRDM16 (R&D, 1:1000), anti-GAPDH (Millipore, 1:3000), or anti-ANGPT2 (R&D, 1:500) antibodies. After washing, membranes were incubated with anti-sheep-HRP (Sigma, 1:1000), anti-mouse-HRP (GE Healthcare, 1:1000), or anti-Goat-HRP (Millipore, 1:5000) secondary antibodies at room temperature for 1.5 h. Membranes were washed and then imaged on a ChemiDoc (Bio-Rad) using SuperSignal West Pico PLUS (ThermoFisher) or combined Pico PLUS and Femto Chemiluminescent Substrates (ThermoFisher, 9:1).
Statistical analysisIB4 stained retinas were quantified in Fiji (ImageJ (Schindelin et al., 2012)). Vascular extension was measured by dividing the angiogenic length by the total retina petal length. Branch points and vascular areas were quantified on a 500 µm × 500 µm image in Angiotool (Zudaire et al., 2011). Used animal numbers or group numbers are described in their respective figure legends. We calculated p-values with unpaired Student’s t-test with Prism9 (GraphPad). A p-value <0.05 was considered to represent a statistically significant difference. Data are presented as mean ± SD.
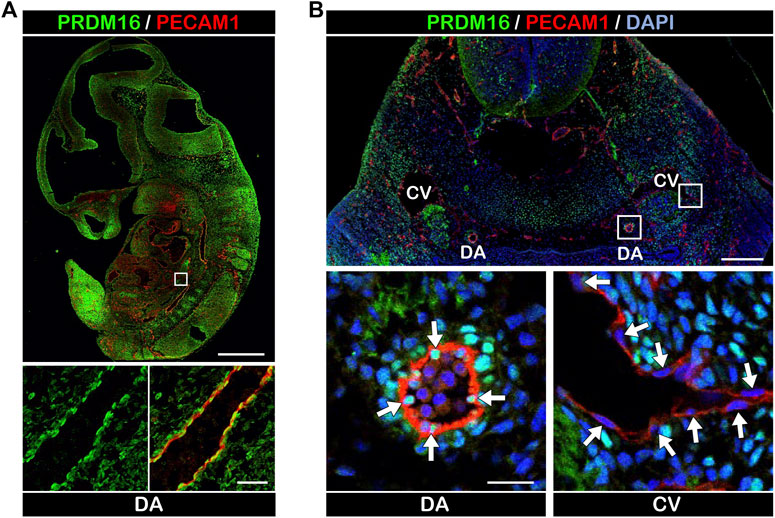
ResultsPRDM16 is expressed in the arterial ECs in developing embryonic vesselsTo investigate the expression of PRDM16 in developing vessels, we performed immunostaining using embryonic day (E) 11.5 wild-type mouse embryos. PRDM16 was detected in several tissues including the heart and somites. Interestingly, PRDM16 was also detected in the PECAM1-positive vascular endothelial cells (ECs) in the dorsal aorta (DA) (Figure 1A). As shown in the transverse section through the pharyngeal arch region, PRDM16 can be detected in the ECs in the DA (arrows in Figure 1B, lower left panel). In contrast, ECs in the cardinal vein (CV) did not show apparent staining of PRDM16 (arrows in Figure 1B, lower right panel). The PRDM16 expression was also observed in the smooth muscle alpha actin (αSMA)-positive vascular smooth muscle cells (vSMCs) (arrow heads in Supplemental Figure S1, lower left panel). These observations suggest that PRDM16 is expressed in the arterial ECs and vSMCs but not in the venous ECs in developing vessels.
FIGURE 1. PRDM16 is expressed in arterial ECs in E11.5 embryos (A) PRDM16 and PECAM1 immunostaining of sagittal section of mouse embryo at E11.5. PRDM16 (green) can be detected in ECs (PECAM1, red) of the dorsal aorta (lower panel). DA, dorsal aorta. Scale bars, 1 mm (upper panel), 50 µm (lower panel). (B) PRDM16 and PECAM1 staining of transverse section of mouse embryo. Higher magnification of DA (left) and CV (right) are shown in the lower panels. DA, dorsal aorta, CV, cardinal vein. Scale bars, 200 µm (upper panel), 20 µm (lower panel).
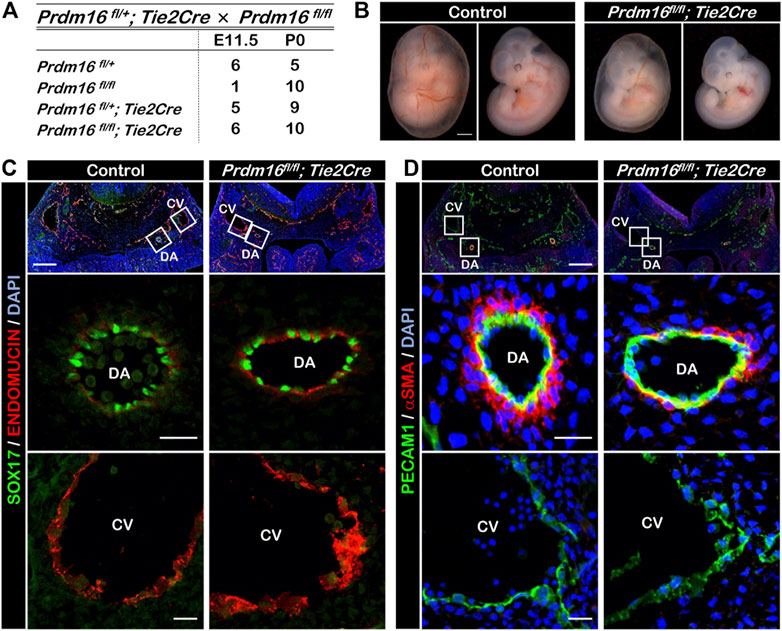
Arterial ECs in the EC-specific Prdm16 knockout embryos display reduction of arterial characteristicsTo investigate the role of PRDM16 in the ECs during development, we bred Prdm16 fl/fl mice with the Tie2Cre mouse line to generate EC-specific Prdm16 knockout embryos (Prdm16 fl/fl; Tie2Cre). The loss of Prdm16 in ECs did not cause any changes to the expected Mendelian ratios at postnatal day (P) 0 (Figure 2A). The vascular formation in the yolk sac of Prdm16 fl/fl; Tie2Cre embryos appeared to be normal at E11.5, but the knockout embryos seemed to be smaller than the control embryos (Figure 2B). We investigated whether deletion of Prdm16 can affect arterial characteristics as we have shown that PRDM16 is specifically expressed in arterial ECs. Immunostaining analysis showed that the expression of the arterial marker SOX17 was comparable in ECs of DA between of the control and Prdm16 mutant embryos (Figure 2C). The venous EC marker, ENDOMUCIN, was expressed only in the CV but not in the DA of control embryos, whereas we observed ectopic expression of ENDOMUCIN in the DA of Prdm16 fl/fl; Tie2Cre embryos (Figure 2C). We next investigated the vSMC recruitment because the arterial ECs start to recruit the vSMCs around E10.5, which is one of the characteristics of arterial ECs. The DAs of control embryos were surrounded by a thick layer of αSMA-positive vSMCs, whereas only a few vSMCs were observed in the DAs of the Prdm16 fl/fl; Tie2Cre embryos (Figure 2D). These results suggest that loss of Prdm16 in developing ECs leads to a decrease of arterial characteristics with increased venous marker expression in the arterial ECs of Prdm16fl/fl; Tie2Cre embryos.
FIGURE 2. Embryonic deletion of Prdm16 in ECs causes ectopic ENDOMUCIN expression and reduced vSMC recruitment to arteries (A) Summary of genotypes of litters obtained by breeding between Prdm16fl/fl female and Prdm16fl/+; Tie2Cre male mice. Genotyping was performed at E11.5 and P0. No dead embryos or mice were noticed. (B) Whole-mount images of E11.5 control and Prdm16fl/fl; Tie2Cre embryos, with and without yolk sac (left and right panels, respectively). Scale bar, 1 mm. (C) Immunostaining of transverse sections of E11.5 control and Prdm16fl/fl; Tie2Cre embryos for SOX17 (green) and ENDOMUCIN (red). Scale bars, 200 µm (upper panels), 20 µm (middle and lower panels). DA, dorsal aorta, CV, cardinal vein. (D) Immunostaining of transverse sections of E11.5 control and Prdm16 fl/fl; Tie2Cre embryos for PECAM1 (green), and αSMA (red). Scale bars, 200 µm (upper panels), 20 µm (middle and lower panels).
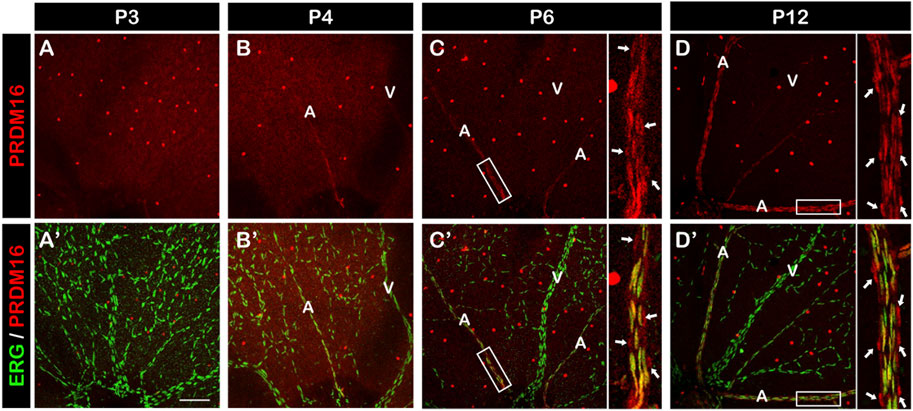
PRDM16 is expressed in arterial ECs of the retinaWe next investigated the role of PRDM16 in postnatal angiogenesis using the mouse retina as a model system. The retinal vessels start to develop at birth, allowing for a time course analysis of vascular formation. At P3, premature retinal vessels were already formed, but we didn’t observe PRDM16 expression in ECs (Figure 3A, A′). By P4, arteriovenous specification has occurred and, interestingly, PRDM16 started to be expressed in the arterial ECs but not in the venous ECs (Figure 3B, B′). From P6, when arterial ECs start to recruit vSMCs, PRDM16 can be detected in both arterial ECs and vSMCs (Figure 3C, C′) and the same expression pattern persisted at a later time point during retinal angiogenesis (Figure 3D, D′) (Jeong et al., 2017). Note that PRDM16 was also expressed in the ganglion cells (bright dots in each retina) (Su et al., 2020). This time course expression analysis indicates that PRDM16 starts to be expressed in the retinal arterial ECs at the beginning of arteriovenous specification.
FIGURE 3. PRDM16 expression in developing mouse retina. Immunostaining of PRDM16 (red) and ERG (green) in whole-mount retina at the indicated time points for P3 (A, A′), P4 (B, B′), P6 (C, C′), and P12 (D, D′). Insets in (C, C′), (D, D′) show high-magnification images of arteries in the P6 and P12 retina. Arrows in D and D′ indicate likely vSMCs surrounding artery that are known to express PRDM16. A: Artery. V: Vein. n = 3. Scale bar, 100 µm. Note that bright red dots throughout retinas are ganglion cells.
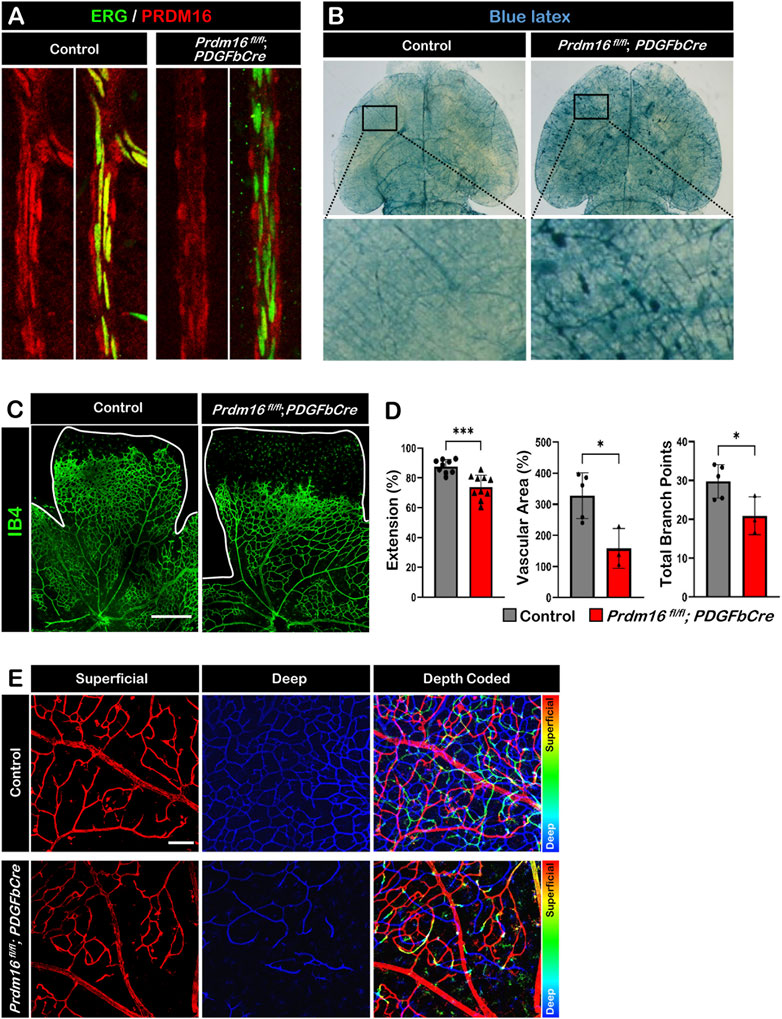
Loss of Prdm16 prevents endothelial migration in developing retinal vesselsTo investigate the role of PRDM16 in the developing vasculature, we generated inducible EC-specific Prdm16 knockout mice (Prdm16 fl/fl; PDGFbCre). Tamoxifen was administered from P1 to P3, and tissues were collected for analysis between P8 and P12. Deletion of PRDM16 in ECs was confirmed by immunostaining, and PRDM16 remained unchanged in vSMCs in Prdm16 fl/fl; PDGFbCre mice at P12 (Figure 4A). To examine the global effect of Prdm16 loss on the vasculature, we injected blue latex into control and Prdm16 fl/fl; PDGFbCre mice at P9. We found abnormal vascular morphology in the brain of Prdm16 fl/fl; PDGFbCre mice compared to the control mice (Figure 4B). To further investigate the role of Prdm16 in angiogenesis and any resulting phenotype, we collected retinas from control and Prdm16 fl/fl; PDGFbCre mice at P8 and stained them with IB4 to visualize the vascular plexus. Although PRDM16 is normally expressed only in the arterial ECs, we found significantly less vascular extension, vascular area, and fewer branch points in the superficial layer of Prdm16 fl/fl; PDGFbCre retinas (Figures 4C, D). In the P12 control retinas, some ECs on the superficial layer migrated into the inner retina and generated the deep retinal vascular plexus (Figure 4E). In Prdm16 fl/fl; PDGFbCre retinas, however, we observed a dramatic decrease of deep retinal vascular plexus formation (Figure 4E). Together, loss of Prdm16 in ECs causes a reduction of vascular migration and disturbed deep plexus formation.
FIGURE 4. Deletion of Prdm16 results in retina vascular defects (A) PRDM16 (red) and ERG (green) co-immunostaining of control and Prdm16fl/fl; PDGFbCre mouse retinas at P12 to show Prdm16 deletion efficiency. (B) Gross image of P9 brains injected with blue latex to visualize vasculature. Insets show normal vasculature in the control mice (left), while vascular abnormalities are seen in the Prdm16fl/fl; PDGFbCre mice (right). (C) Whole mount IB4 staining of P8 control and Prdm16fl/fl; PDGFbCre retina. n > 3. Scale bar, 500 µm. (D) Quantification of the vasculature extension (control n = 9, Prdm16fl/fl; PDGFbCre n = 10), vascular area (control n = 5, Prdm16fl/fl; PDGFbCre n = 3), and total branch points (control n = 5, Prdm16fl/fl; PDGFbCre n = 3). Data are as mean ± SD, *: p < 0.05, ***: p < 0.001 (Student’s t-test). (E) Representative pseudo-colored images of P12 control and Prdm16fl/fl; PDGFbCre mouse retinas. Vasculature marked with IB4. Superficial and deep plexus images were overlaid using ImageJ, superficial layer in red, changing to blue moving to the deep plexus. Scale bar, 100 µm, n = 3.
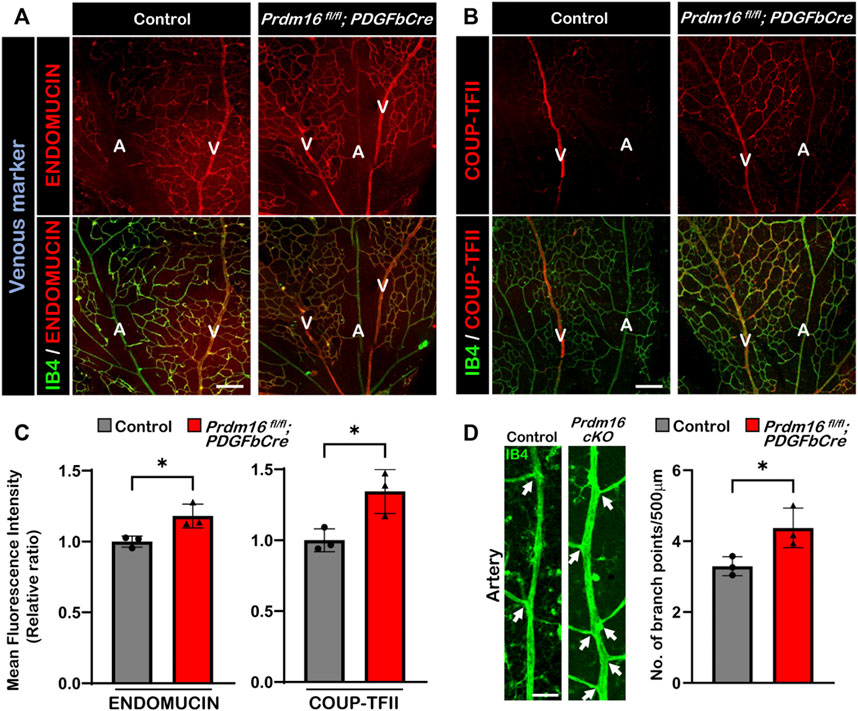
Loss of Prdm16 in ECs leads to ectopic venous marker expression in the retinal arteriesAs we observed ectopic ENDOMUCIN expression in the DAs of Prdm16 fl/fl; Tie2Cre embryos, we next investigated whether venous marker expression was also increased in arterial ECs in Prdm16 fl/fl; PDGFbCre retinas. We found ectopic ENDOMUCIN and COUP-TFII expression in the arterial ECs of Prdm16 fl/fl; PDGFbCre retinas (Figures 5A–C). In line with the ectopic venous marker expression, the Prdm16 fl/fl; PDGFbCre mice showed more branches in the arteries than in control mice, indicating a slight shift towards being more vein like (Figure 5D). To examine if loss of Prdm16 altered the expression of artery markers, we performed immunostaining for SOX17 and RT-qPCR using RNA from P9 whole retina. No changes were found in SOX17 or the artery markers such as Cxcr4, Dll4, or Hrt2 (Supplementary Figures S2A, S2B). Since the phenotypes of the arterial ECs in Prdm16 fl/fl; PDGFbCre retinas are consistent with Prdm16 fl/fl; Tie2Cre embryos, these results strongly suggest that PRDM16 is necessary for proper vascular development and may be involved in regulating arterial specification during the vascular formation.
FIGURE 5. Ectopic expression of venous markers ENDOMUCIN and COUP-TFII in Prdm16fl/fl; PDGFbCre arteries. Immunostaining of whole mount P9 retina with IB4 (green) and venous markers in red (ENDOMUCIN (A) or COUP-TFII (B)). Scale bar, 100µm, n = 3. (C) Quantification of mean fluorescence intensity of ENDOMUCIN and COUP-TFII in Prdm16fl/fl; PDGFbCre mice normalized to the control retinas. n = 3; Data are as mean ± SD, Student’s t-test: *p < 0.05. (D) Representative images of P9 retinal arteries in control and Prdm16fl/fl; PDGFbCre (Prdm16 cKO) mice (left panel) and quantification of the number of branch points (right panel). Arrows indicate branch points. n = 3; Data are as mean ± SD, Student’s t-test: *p < 0.05.
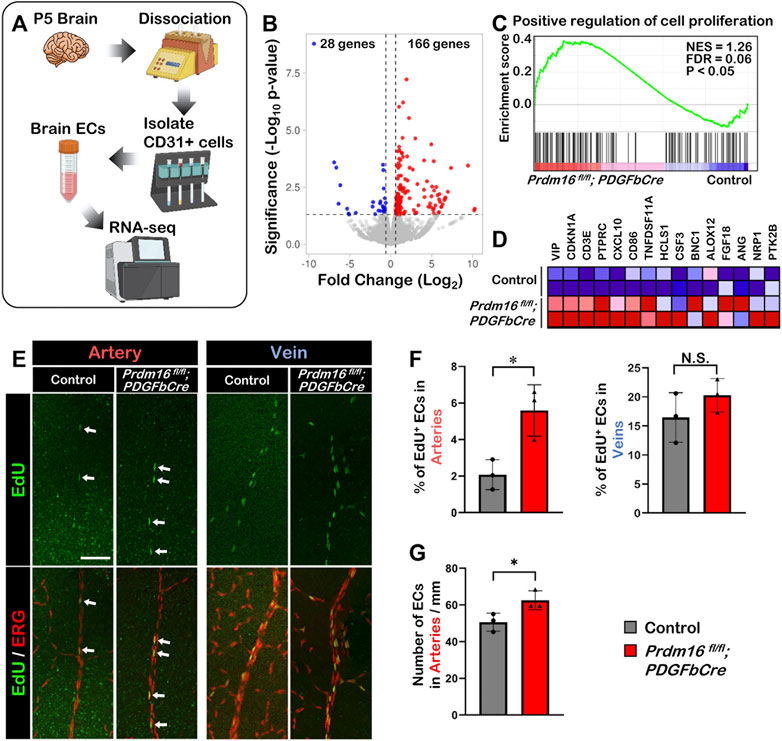
Loss of Prdm16 causes more proliferation in retinal arterial ECsTo investigate the molecular function of PRDM16 during vascular development, we performed RNA-seq analysis using isolated ECs from P5 brains (Figure 6A). The volcano plot of the differential gene expression analysis showed that 166 genes were upregulated, and 28 genes were downregulated significantly in the Prdm16 fl/fl; PDGFbCre ECs compared with those of littermate controls (Figure 6B). Since we observed ectopic venous marker expression in arterial ECs of Prdm16 fl/fl; PDGFbCre retinas, we hypothesized that Prdm16 KO arterial ECs may show some venous characteristics. We first focused on cell proliferation because EC proliferation in veins is generally higher than in arteries (McDonald et al., 2018; Sabbagh et al., 2018). GSEA analysis using a gene set for the “positive regulation of cell proliferation” showed that genes related to cell proliferation were significantly enriched in the Prdm16 fl/fl; PDGFbCre brain ECs (Figures 6C, D). To examine the EC proliferation in the developing retinal vessels, 5-ethynyl-2′-deoxyuridin (EdU) was administered to P7 mice via IP injection 24 h before analysis. In contrast to only a few EdU-positive ECs detected in the arteries of control retinas, there were more EdU-positive arterial ECs in Prdm16 fl/fl; PDGFbCre mice (Figures 6E, F). Consistently, we also observed an increased number of ECs in the arteries of the Prdm16fl/fl; PDGFbCre mice as compared with the control mice (Figure 6G). Note that there was no significant change of venous EC proliferation between control and Prdm16fl/fl; PDGFbCre retinas (Figures 6E, F), suggesting that PRDM16 might suppress the endothelial proliferation in the arteries during retinal vascular formation.
FIGURE 6. Loss of Prdm16 leads to increased proliferation in arterial endothelial cells (A) Schematic for isolation of brain ECs from P5 mouse brains for RNA-seq. (B) Volcano plot of differentially expressed genes from P5 Prdm16fl/fl; PDGFbCre brain ECs. Blue dots represent downregulated genes, while red dots represent upregulated genes. Gray dots failed to meet minimum fold change of 1.5 or were not differentially expressed. (C) GSEA plot shows that cell proliferation genes were upregulated in Prdm16fl/fl; PDGFbCre ECs. (D) Heat map for top 15 genes in gene set. (E) Whole mount EdU staining of P8 retina. EdU (green) to label proliferating cells, ERG (red) used to mark EC nuclei. Scale bar, 100 µm. (F) Quantification of EdU+ ECs in arteries. Counted total number of EdU and ERG double positive cells in artery trunks until 50% extension, normalized to total number ERG single positive cells in the counted area. n = 3; Data are as mean ± SD, Student’s t-test: *p < 0.05. (G) Quantification of total number of ECs/mm in control and Prdm16fl/fl; PDGFbCre mouse retinal arteries. n = 3; Data are as mean ± SD, Student’s t-test: *p < 0.05.
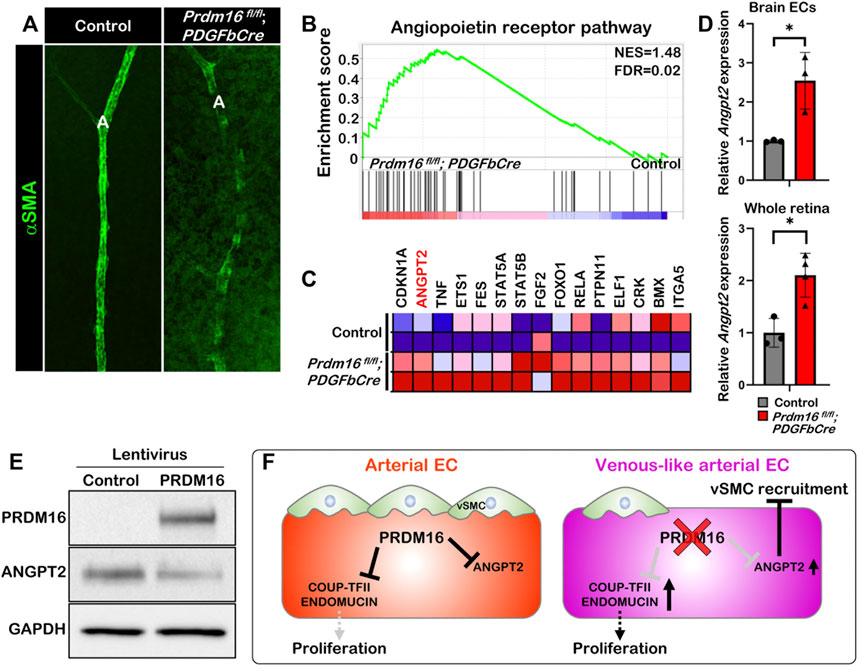
Loss of Prdm16 induces Angpt2 expression in ECsThe ectopic venous marker expression in the arterial ECs of Prdm16 mutant mice led us to further examine whether postnatal loss of Prdm16 in ECs leads to a reduced capability to recruit vSMCs to arteries in the retina, as seen in the DAs of Prdm16fl/fl; Tie2Cre embryos. The immunostaining for αSMA showed less vSMC recruitment in the Prdm16fl/fl; PDGFbCre arteries (Figure 7A). To investigate a potential mechanism for the lack of vSMC recruitment seen in the Prdm16fl/fl; Tie2Cre embryos and Prdm16fl/fl; PDGFbCre retinas, we analyzed our RNA-seq data. Interestingly, GSEA analysis using a gene set for the angiopoietin receptor pathway showed that genes related to the angiopoietin pathway were significantly enriched in the Prdm16fl/fl; PDGFbCre brain ECs (Figures 7B, C). Furthermore, Angpt2 was among the top 15 genes upregulated in the Prdm16fl/fl; PDGFbCre brain ECs as shown in the heatmap (Figure 7C). To confirm the upregulation of Angpt2 expression, we performed RT-qPCR analysis using RNA from P5 isolated brain ECs or P9 whole retinas. The data showed that the expression of Angpt2 was significantly increased in Prdm16fl/fl; PDGFbCre brain ECs and retinas (Figure 7D), suggesting that PRDM16 may act as a repressor for Angpt2 expression in arterial ECs. Conversely, forced expression of PRDM16 in HUVECs was sufficient to drive arterial gene expression in the venous ECs (Supplementary Figure S3) and reduce the ANGPT2 level (Figure 7E). Together, these results suggest that PRDM16 promotes vSMC recruitment through suppression of Angpt2 expression in arterial ECs (Figure 7F).
FIGURE 7. PRDM16 represses Angpt2 expression in ECs (A) Whole mount αSMA immunostaining of control and Prdm16fl/fl; PDGFbCre retina at P9. αSMA, alpha smooth muscle actin (green). (B) GSEA plot shows that angiopoietin receptor pathway was upregulated in Prdm16fl/fl; PDGFbCre ECs. (C) Heat map of the top 15 dysregulated genes in Angiopoietin receptor pathway. (D) RT-qPCR analysis of Angpt2 expression in P5 isolated brain ECs and P9 whole retina lysates. n = 3; Data are as mean ± SD, Student’s t-test: *p < 0.05. (E) Western blot of cell lysates from HUVECs infected with lentivirus expressing PRDM16 using antibodies against PRDM16, ANGPT2, and GAPDH. (F) Model for the role of PRDM16 in the maintenance of arterial EC characteristics. PRDM16 represses proliferation and the expression of venous markers in arterial ECs and inhibits Angpt2 in arterial ECs, which allows vSMCs to associate to the arteries. Loss of PRDM16 leads to an increase in the proliferation of arterial ECs and the expression of venous markers and Angpt2 leading to a reduction in vSMC recruitment to the arteries.
DiscussionIn the present study, we investigated the role of PRDM16 in ECs during vascular development. We found that loss of PRDM16 induced ectopic venous marker expression, less vSMC recruitment, and promoted cell proliferation in the arterial ECs. We also found that the Prdm16 KO arterial ECs showed elevation of Angpt2 expression that would prevent vSMC recruitment. These results strongly suggest that PRDM16 may play an important role in maintaining arterial characteristics.
At the beginning of vascular formation, immature ECs form the primitive vascular network, then arterial-venous specification occurs to mature the vessels (Swift and Weinstein, 2009). During the arterial-venous specification, immature ECs start to express arterial or venous markers. It is well known that the VEGF-Notch signaling pathway promotes arterial specific gene expression leading ECs towards an arterial fate (Corada et al., 2013). Conversely, the chicken ovalbumin upstream promoter transcription factor II (COUP-TFII/Nr2f2) has been reported to be expressed in venous ECs (Corada et al., 2013). ENDOMUCIN (encoded by Emcn) is an O-glycosylated single transmembrane sialomucin that is highly expressed in venous and capillary ECs, but not in the arterial ECs (Morgan et al., 1999; Kuhn et al., 2002). ENDOMUCIN was identified as a VEGF-induced angiogenic regulator (Park-Windhol et al., 2017). A previous in vitro study showed that loss of ENDOMUCIN function induces less EC migration, proliferation, and tube formation, while its over-expression enhances these phenotypes (Park-Windhol et al., 2017). We found that knockout of Prdm16 in the ECs promotes ectopic ENDOMUCIN expression in the arterial ECs. Normally, arterial ECs in the retina show less proliferation while more proliferation is observed in venous ECs. Here we observed in the arterial ECs of Prdm16 knockout retinas showed more proliferation compared to the control retinas, suggesting the possibility that the ectopic ENDOMUCIN expression may contribute to the increased cell proliferation in the Prdm16 knockout arterial ECs.
We observed ectopic ENDOMUCIN and COUP-TFII expression in the Prdm16 KO arterial ECs, but it remains unclear whether PRDM16 directly regulates Emcn or Nr2f2 expression during vascular development. We predict that PRDM16 doesn’t have a strong ability to completely change the arterial-venous fate, but that it could be involved in the molecular mechanisms that maintain arterial characteristics. To elucidate the PRDM16 function during vascular formation, genome-wide chromatin immunoprecipitation sequencing (ChIP-seq) analysis is required.
Following the arterial-venous specification, arterial ECs start to recruit mural cells, such as pericytes and vSMCs, to stabilize vascular structure to sustain the arterial function (Gaengel et al., 2009). If the vSMC recruitment is compromised, blood vessels might become dilated and tend to be leaky (Hellstrom et al., 1999; Qin et al., 2013). It has been well characterized that the platelet-derived growth factor-B (PDGF-B) and the angiopoietins regulate paracrine interactions between ECs and vSMCs (Bergers and Song, 2005). Knockout of Angpt1 (encoding ANGIOPOIETIN1) or its receptor Tek (encoding TIE2) induces embryonic lethality due to the failure of ECs to associate with the adjacent vSMCs. Transgenic mice overexpressing Angpt2 showed a similar phenotype of Angpt1 or Tek knockout embryos, indicating that ANGIOPOIETIN2 antagonizes TIE2 and competes for binding at the receptor with ANGIOPOIETIN1 (Maisonpierre et al., 1997). In this study, we found that Angpt2 expression was upregulated in the arterial EC of Prdm16 knockout retinas. Interestingly, consistent with Angpt2 transgenic mice, Prdm16 knockout embryos and retinas showed less vSMC recruitment to arterial ECs. To investigate whether PRDM16 directly regulates Angpt2 expression, we examined available PRDM16 ChIP-seq data from heart ventricles (Wu et al., 2022) and found a peak upstream of the Angpt2 transcription start site, strongly suggesting that PRDM16 promotes vSMC recruitment through the inhibition of Angpt2 expression.
Although PRDM16 is only expressed in the arterial ECs, the Prdm16 knockout retinas displayed less vascular formation entirely. It is also known that ANGIOPOIETIN1-TIE2 signaling pathway induces endothelial cell sprouting through the phosphatidylinositol 3′-kinase (PI3K)-AKT signaling pathway, and ANGIOPOIETIN2 disrupts vascular formation in the developing embryos (Maisonpierre et al., 1997; Morales-Ruiz et al., 2000). Since Prdm16 knockout retinas showed upregulation of ANGPOIETIN2 expression, we hypothesize that paracrine ANGIPOIETIN2 signaling may also affect the neighboring PRDM16-negative ECs during vascular development. Deeper analysis for the activity of PI3K/AKT signaling pathway in the Prdm16 knockout ECs is required to elucidate this question.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/946755.
Ethics statementThe animal study was reviewed and approved by the Institutional Animal Care and Use Committee of Cincinnati Children's Hospital Medical Center.
Author contributionsMT: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing. MS: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing. MV: Validation. JH: Resources. SM: data acquisition. QRL: Resources, supervision, validation. MX: Conceptualization, resources, formal analysis, supervision, funding acquisition, validation, investigation, writing, project administration. All authors contributed to the article and approved the submitted version
FundingThis work was supported in part by grants from the National Institute of Health (HL132211, S105787 and EY032029).
AcknowledgmentsThe authors are grateful to Dr. Elisa Boscolo for her insightful discussions and suggestions and Dr. Zaili Luo for his assistance in lentivirus generation.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1165379/full#supplementary-material
ReferencesAguilo, F., Avagyan, S., Labar, A., Sevilla, A., Lee, D. F., Kumar, P., et al. (2011). Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood 117 (19), 5057–5066. doi:10.1182/blood-2010-08-300145
PubMed Abstract | CrossRef Full Text | Google Scholar
Becerril, S., Gomez-Ambrosi, J., Martin, M., Moncada, R., Sesma, P., Burrell, M. A., et al. (2013). Role of Prdm16 in the activation of Brown fat programming. Relevance to the development of obesity. Histol. Histopathol. 28 (11), 1411–1425. doi:10.14670/HH-28.1411
PubMed Abstract | CrossRef Full Text | Google Scholar
Biferali, B., Bianconi, V., Perez, D. F., Kronawitter, S. P., Marullo, F., Maggio, R., et al. (2021). Prdm16-Mediated H3k9 methylation controls fibro-adipogenic progenitors identity during skeletal muscle repair. Sci. Adv. 7 (23). Epub 2021/06/04. doi:10.1126/sciadv.abd9371
CrossRef Full Text | Google Scholar
Birjiniuk, A., Rosenfeld, J., Tunuguntla, H., Allen, H., Penny, D., Kim, J., et al. (2018). Deletions and loss of function mutations in Prdm16 are associated with pediatric cardiomyopathy. Circulation 138, 12162. doi:10.1161/circ.138.suppl_1.12162
CrossRef Full Text | Google Scholar
Bjork, B. C., Gomez, A. C., Ahmed, A., Aumann, M., Jones, J., Saadi, I., et al. (2018). Prdm16 and mecom mutants exhibit cleft secondary palate as a result of perturbations that affect different stages of palatogenesis. Faseb J. 32 (1), 776.7. doi:10.1096/fasebj.2018.32.1_supplement.776.7
CrossRef Full Text | Google Scholar
Bjork, B. C., Turbe-Doan, A., Prysak, M., Herron, B. J., and Beier, D. R. (2010). Prdm16 is required for normal palatogenesis in mice. Hum. Mol. Genet. 19 (5), 774–789. doi:10.1093/hmg/ddp543
PubMed Abstract | CrossRef Full Text | Google Scholar
Borensztein, M., Viengchareun, S., Montarras, D., Journot, L., Binart, N., Lombes, M., et al. (2012). Double myod and Igf2 inactivation promotes Brown adipose tissue development by increasing Prdm16 expression. Faseb J. 26 (11), 4584–4591. doi:10.1096/fj.12-208496
PubMed Abstract | CrossRef Full Text | Google Scholar
Chen, W., He, S., and Xiang, D. (2021). Hypoxia-induced retinal pigment epithelium cell-derived bfgf promotes the migration and angiogenesis of huvecs through regulating tgf-ß1/smad2/3 pathway. Gene 790, 145695. Epub 20210505. doi:10.1016/j.gene.2021.145695
PubMed Abstract | CrossRef Full Text | Google Scholar
Chuikov, S., Levi, B. P., Smith, M. L., and Morrison, S. J. (2010). Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat. Cell Biol. 12 (10), 999–1006. Epub 2010/09/14. doi:10.1038/ncb2101
PubMed Abstract | CrossRef Full Text | Google Scholar
Cibi, D. M., Bi-Lin, K. W., Shekeran, S. G., Sandireddy, R., Tee, N., Singh, A., et al. Prdm16 deficiency leads to age-dependent cardiac hypertrophy, adverse remodeling, mitochondrial dysfunction, and heart failure. Cell Rep. (2020) 33(3). doi:10.1016/j.celrep.2020.108288
PubMed Abstract | CrossRef Full Text | Google Scholar
Claxton, S., Kostourou, V., Jadeja, S., Chambon, P., Hodivala-Dilke, K., and Fruttiger, M. (2008). Efficient, inducible cre-recombinase activation in vascular endothelium. Genesis 46 (2), 74–80. Epub 2008/02/08. doi:10.1002/dvg.20367
PubMed Abstract | CrossRef Full Text | Google Scholar
Cohen, P., Levy, J. D., Zhang, Y. Y., Frontini, A., Kolodin, D. P., Svensson, K. J., et al. (2014). Ablation of Prdm16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156 (1-2), 304–316. doi:10.1016/j.cell.2013.12.021
PubMed Abstract | CrossRef Full Text | Google Scholar
Corada, M., Orsenigo, F., Morini, M. F., Pitulescu, M. E., Bhat, G., Nyqvist, D., et al. (2013). Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 4, 2609. Epub 2013/10/25. doi:10.1038/ncomms3609
PubMed Abstract | CrossRef Full Text | Google Scholar
Corrigan, D. J., Luchsinger, L. L., de Almeida, M. J., Williams, L. J., Strikoudis, A., and Snoeck, H. W. (2018). Prdm16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J. Clin. Investigation 128 (8), 3250–3264. doi:10.1172/Jci99862
PubMed Abstract | CrossRef Full Text | Google Scholar
Craps, S., Van Wauwe, J., De Moudt, S., De Munck, D., Leloup, A. J. A., Boeckx, B., et al. (2021). Prdm16 supports arterial flow recovery by maintaining endothelial function. Circulation Res. 129 (1), 63–77. doi:10.1161/Circresaha.120.318501
PubMed Abstract | CrossRef Full Text | Google Scholar
Crist, A. M., Young, C., and Meadows, S. M. (2017). Characterization of arteriovenous identity in the developing neonate mouse retina. Gene Expr. Patterns 23-24, 22–31. Epub 2017/02/09. doi:10.1016/j.gep.2017.01.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Cunha, S. I., Magnusson, P. U., Dejana, E., and Lampugnani, M. G. (2017). Deregulated tgf-Β/bmp signaling in vascular malformations. Circulation Res. 121, 981–999. doi:10.1161/CIRCRESAHA.117.309930
PubMed Abstract | CrossRef Full Text | Google Scholar
Dumont, D. J., Gradwohl, G., Fong, G. H., Puri, M. C., Gertsenstein, M., Auerbach, A., et al. (1994). Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, Tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 8 (16), 1897–1909. doi:10.1101/gad.8.16.1897
PubMed Abstract | CrossRef Full Text | Google Scholar
Fischer, A., Schumacher, N., Maier, M., Sendtner, M., and Gessler, M. (2004). The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 18 (8), 901–911. doi:10.1101/gad.291004
PubMed Abstract | CrossRef Full Text | Google Scholar
Fog, C. K., Galli, G. G., and Lund, A. H. (2012). Prdm proteins: Important players in differentiation and disease. Bioessays 34 (1), 50–60. Epub 2011/10/27. doi:10.1002/bies.201100107
PubMed Abstract | CrossRef Full Text | Google Scholar
Gaengel, K., Genové, G., Armulik, A., and Betsholtz, C. (2009). Endothelial-mural cell signaling in vascular development and angiogenesis. Arteriosclerosis, Thrombosis, Vasc. Biol. 29, 630–638. doi:10.1161/ATVBAHA.107.161521
PubMed Abstract | CrossRef Full Text | Google Scholar
Gale, N. W., Thurston, G., Hackett, S. F., Renard, R., Wang, Q., McClain, J., et al. (2002). Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by angiopoietin-1. Dev. Cell 3 (3), 411–423. doi:10.1016/s1534-5807(02)00217-4
PubMed Abstract | CrossRef Full Text | Google Scholar
Gan, L., Liu, Z., Feng, F., Wu, T., Luo, D., Hu, C., et al. (2018). Foxc2 coordinates inflammation and browning of white adipose by leptin-stat3-prdm16 signal in mice. Int. J. Obes. 42 (2), 252–259. doi:10.1038/ijo.2017.208
PubMed Abstract | CrossRef Full Text | Google Scholar
Gerhardt, H., Golding, M., Fruttiger, M., Ruhrberg, C., Lundkvist, A., Abramsson, A., et al. (2003). Vegf guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161 (6), 1163–1177. Epub 2003/06/16. doi:10.1083/jcb.200302047
PubMed Abstract | CrossRef Full Text | Google Scholar
Harms, M. J., Ishibashi, J., Wang, W. S., Lim, H. W., Goyama, S., Sato, T., et al. (2014). Prdm16 is required for the maintenance of Brown adipocyte identity and function in adult mice. Cell Metab. 19 (4), 593–604. doi:10.1016/j.cmet.2014.03.007
PubMed Abstract | CrossRef Full Text | Google Scholar
Hellstrom, M., Kal, N. M., Lindahl, P., Abramsson, A., and Betsholtz, C. (1999). Role of pdgf-B and pdgfr-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 126 (14), 3047–3055. doi:10.1242/dev.126.14.3047
PubMed Abstract | CrossRef Full Text | Google Scholar
Jeong, H. W., Hernández-Rodríguez, B., Kim, J., Kim, K. P., Enriquez-Gasca, R., Yoon, J., et al. (2017). Transcriptional regulation of endothelial cell behavior during sprouting angiogenesis. Nat. Commun. 8 (1), 726. Epub 20170928. doi:10.1038/s41467-017-00738-7
PubMed Abstract | CrossRef Full Text | Google Scholar
Kisanuki, Y. Y., Hammer, R. E., Miyazaki, J., Williams, S. C., Richardson, J. A., and Yanagisawa, M. (2001). Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230 (2), 230–242. doi:10.1006/dbio.2000.0106
留言 (0)