
記住我








Many proteins are characterized by a given three-dimensional structure when they are observed in a water soluble monomeric state Branden and Tooze (1999). In order to understand the way the sequence determines the structure, the effect of single point mutations has been studied since a long time Cunningham and Wells (1989). A simple way to address the sequence-structure interplay is to measure some structural parameter as a function of temperature. Circular dichroism (CD) and many other techniques are often used and in many cases the change of this structural parameter with temperature can be taken as an indicator of the melting of the protein structure Cantor and Schimmel (1980). The change of melting temperature triggered by different single point mutations is therefore a widely used measure of the change of protein stability upon a localized change of the protein sequence and large archives of such data have been collected Guerois et al. (2002); Alexov and Sternberg (2013); Forbes et al. (2016). When this information is available, it must be interpreted in terms of the reshaping of atomic interactions.
The change of protein stability upon sequence mutations has implications in many pathologies. One example is Friedreich’s ataxia, an autosomal-recessive genetic condition that causes ataxia, sensory loss and cardiomyopathy worsening over time Pandolfo (2009); Klockgether (2011); Clark et al. (2018). The cause of the disease is in mutations of the gene encoding for the frataxin (FXN) protein. Depending on the specific kind of mutation, a patient may end up with an insufficient level of frataxin, a nonfunctional frataxin or frataxin, that is, not correctly localized in the mitochondria Delatycki et al. (2000); Galea et al. (2016). Frataxin variants have also a role in cancer, as expected because of the involvement of FXN and mitochondria in the control of oxidative metabolism Schulz et al. (2006). Indeed, missense variants are found in multiple human cancer tissues Petrosino et al. (2019, 2021). The example of FXN shows that even single point mutations can have significant impact in protein stability, trafficking, plasticity, interactions with local environment and mutual interactions with other macromolecules.
Many models have been proposed to relate measured changes in protein stability with the chemical nature of the protein sequence. High through-put approaches based on atomic models have been recently developed Steinbrecher et al. (2017). Many of these approaches are summarized in this Special Issue.
The method we would like to propose here aims at predicting the change of thermal stability of a protein in a monomeric water-soluble state, when its sequence is changed by a single aminoacid. The method is based on a suitable modelling of interatomic forces, i.e., it is atomistic, and includes an explicit model of the water solution. The method was initially applied to FXN Botticelli et al. (2022) and it is here refined and discussed in more detail, to achieve better computational performance and higher accuracy in prediction. In particular, we use here the well-tempered metadynamics, one of the best performing method to enhance sampling of phase space in atomistic models. A small reference protein of 58 residues is first used to assess the methods and to understand limitations and advantages.
Many initial configurations of the protein of interest are generated, assuming the protein structure representing the native state of the wild-type sequence, but with initial conditions diversified as much as possible. This is achieved by changing the protein environment, that is, in this case the water solution of NaCl. A multiple walkers metadynamics simulation is carried out Raiteri et al. (2006); Hošek et al. (2016); Hošek et al. (2017), building an external biasing potential as a function of a suitably chosen collective variable. In the range of values spanned by the collective variable folded and unfolded protein structures are sampled.
The external potential is built so as to initially unfold the protein structure. All along the metadynamics simulation time, the external biasing potential is systematically constructed and updated to uniformly sample folded and unfolded configurations. This goal is best achieved by combining multiple walkers histories into a unique trajectory Hošek et al. (2017).
The standard analysis of multiple walkers metadynamics can be performed, but limitations in predicting experimental behaviours arise because of the huge number of configurations required to achieve a good convergence and stability. We therefore decided to exploit the features of the maximal constrained entropy approach which allows to properly re-modulate the collected set of sampled configurations by imposing suitable conformational constraints. In this way we can reliably monitor the change in free energy as a function of average values of the chosen collective variables. The quantity of interest to be calculated is therefore:
ΔΔG=ΔGX−ΔG0=GXs−GXs0−G0s−G0s0,(1)where the subscript denotes a sequence (the pedix 0 indicates a reference sequence, usually the wild-type), and the s state variable indicates the degree of structural order, where s0 indicates the folded native state and s indicates the unfolded state.
The approach is first applied to the study of a paradigmatic protein displaying a well defined three-dimensional structure, namely, to the bovine pancreatic trypsine inhibitor (BPTI), where the effect of a set of mutations on melting temperature has been carefully investigated Yu et al. (1995). BPTI has always been a milestone for folding studies, being one of the smallest proteins (58 aminoacids) characterized by a well defined structure. Then, the same procedure is applied to frataxin, in a truncated form of 121 aminoacids, and 5 of its variants Petrosino et al. (2019).
2 Materials and methodsThe computational methods used in this work are similar to those used in Ref. (Botticelli et al., 2022). In the following we emphasize the differences that characterize this work.
2.1 MetadynamicsLet ξ(q) be a collective variable (CV) function of atomic positions, q. When ξ is an observable quantity, the values, s, allowed for ξ can be used to label system macrostates. The set of coordinates q labels the system microstates, each set of q yielding one of the possible values of s. If ergodicity holds, infinitely long simulations of a trajectory q(t) in a given statistical ensemble would correctly sample the statistical weight of ξ. However, because of the huge number of ways in which certain values of s of ξ are encountered, compared to others, actual numerical simulations in practice only sample the maximally degenerate values of ξ. This is precisely the case where ξ is the CV associated to folding/unfolding events.
Standard statistical ensembles and more recently generalized ensembles try to address this problem by biasing the trajectory to spend more time where ξ has a low degeneracy and less time where ξ has a large degeneracy.
The sampling of configurations obtained with the biased inverse probability of ξ is called metastatistics. We will denote by P̃(q) the probability of microstates encountered along the simulated trajectory and by P̃(ξ) the probability of the macrostates labeled by ξ. For simplicity with a little abuse of notation we use the same name for the metastatistics probability as function of the microscopic variables, q, and to the associated metastatistics probability as function of the macroscopic collective variable, ξ.
Many methods have been proposed to sample configurations with the inverse of the estimated probability of ξ Mitsutake et al. (2001). In this work and in the previous application of the method Botticelli et al. (2022), we used the altruistic metadynamics proposed in Refs. Hošek et al. (2016); Hošek et al. (2017). The desired metastatistics is obtained from a swarm of trajectories provided by metadynamics after building a suitable external bias, which is then kept fixed when collecting configurations in the final step of the NpT simulation (see Section 2.6). We performed simulations in the statistical ensemble associated to constant temperature T and pressure p (NpT ensemble) because macromolecules forced by an external bias undergo large and fast conformational changes. When these conditions occur, solute macromolecules exert strong perturbations over the explicit solvent and ions representing their environment. To cope with steep changes of kinetic energy of water molecules and possible temporary voids around the macromolecule, the NpT ensemble is recommended.
In the framework of metadynamics, the estimated probability of the CV is expressed by means of a sum of Gaussian functions, VG [ξ(q)], related to the inverse metastatistics probability by the formula
with β = 1/(kBT) where T is the temperature used in the simulation, kB the Boltzmann constant, and C a normalization constant, that is, of no relevance in the computation of thermal averages. Different methods have been proposed to build an external bias VG [ξ(q)] such that the probability distribution of ξ is flat and transitions between folded and unfolded states of a biomolecule endowed with many degrees of freedom, are equally well sampled.
In metadynamics the external potential VG (ξ, t) acting on the system at time t is defined as:
VGξq,t=w∑t′=τG,2τG,…exp−ξq−st′22δ2(3)where t′ < t, s(t) = ξ(q(t)) is the value taken by the CV at time t, w is the Gaussian height, δ is the Gaussian width τG is the time interval after which a new Gaussians is added.
After a sufficiently long time VG(s, t) provides an estimate of the underlying free energy F according to the formula
where C(t) depends on time but not on the collective variables s, VG is the external biasing potential acting on the system at time t.
Equation above states that an equilibrium quantity, like free energy, can be estimated by a non-equilibrium dynamics in which the bias potential is changed in time, as new Gaussians are successively added. In metadynamics, when all the wells in CV distribution are filled with Gaussians, the dynamics in the CV space becomes diffusive.
2.2 Well tempered metadynamicsWell tempered metadynamics is an improved approach designed to obtain a reliable estimator of the free energy Barducci et al. (2008). The weight of each Gaussian function added to the bias VG depends on the history of VG (VG(t′)). Equation 3 changes into:
VGξq,t=w∑t′=τG,2τG,…exp−VGξq,t′kBΔT×exp−ξq−st′22δ2,(5)where kBΔT is approximately the energy change when a new value of ξ is visited. An exact relation between VG(s, t) and F(s) can be obtained if the rate at which the bias potential is modified is suitably decreased as the simulation progresses. With well tempered metadynamics, the biasing potential converges to
VGs,t=−ΔTT+ΔTFs+Ct.(6)The quantity T+ΔTT is called “biasing factor”.
For a finite T, the probability distribution is proportional to:
exp−FskBTexpΔTT+ΔTFskBT=exp−FskBT+ΔT(7)which corresponds to effectively increasing the CV sampling temperature. Thus, the effect of well tempered metadynamics is similar to that of other non-equilibrium methods, like steered molecular dynamics, but trajectories are obtained with a quasi-equilibrium procedure Bussi et al. (2018).
In well tempered metadynamics, as the simulation proceeds the width of the added Gaussian remains constant but its height decreases (see Eq. 5). The bias, which increases monotonically, eventually changes very slowly with time. At the beginning the space of CV is flooded by Gaussians of height w. With the progress of flooding, heights of newly added Gaussians decrease. This behaviour is very important in highly complex biological systems, where the bias potential should never reach any excessively large value.
In contrast with the “non tempered” metadynamics, in the well tempered metadynamics a flat CV distibution is not expected to be achieved when convergence is obtained. A simple interpretation of the fact that the distribution of the CV at long times is not flat is the following. Since the prefactor for the accumulated Gaussians depends on the value of s, Gaussians of different heights are placed in different regions of the CV space. In order to reach a stationary distribution, it is thus necessary that the system spends more time in regions where small Gaussians are added and less time in regions where large Gaussians are added. This idea can be pushed further and used to convert metadynamics in an algorithm, not designed to flatten completely (as in non-tempered metadynamics) or partially (as in well tempered metadynamics) the histogram of the CVs but rather to enforce a predefined distribution Bussi et al. (2018).
In this work we used a biasing factor of 20 (see Eq. 6), corresponding to ΔT = 5700 K, in agreement with the biasing factor used in literature for similar molecular systems Hošek et al. (2017). The energy value RΔT is of the order of a typical energy barrier of a single hydrogen bond.
2.3 The maximal constrained entropy methodThe maximal constrained entropy method (MEC method, hereafter) allows, starting from P̃(ξ) of Eq. 2, to obtain a better probability for thermal average calculations. This elaboration is used to correct for limitations of P̃(ξ), whatever the method used for its determination is. We remind that the method consists in post-processing the biased statistics (that we indicate with metastatistics) collected by whatever method. Since in actual simulations one works with trajectories where configurations can be enumerated, we attach the microstate index γ to the configuration and we denote by P̃γ the probability
P̃γ=w̃γ∑γw̃γ,(8)where w̃γ is the number of microstates with label γ collected in the metastatistics and Z̃=∑γw̃γ is a normalization factor.
In an infinitely long (ergodic) simulation, it is unnecessary to explicitly evaluate the weights w̃γ, as they are automatically encoded in the degeneracy of the set of collected configurations sampled along the simulated trajectory. This means that in the following equations, where the sum over γ is extended over that actually produced configurations, we should not include the factor P̃γ. However, we leave this redundant factor to recall that we are dealing with a finite set of configurations generated by metadynamics.
In case of the “non tempered” metadynamics, the maximal constrained entropy was employed as a viable solution to compute thermal averages as a function of the average values taken by the chosen CV, in situations where metastatistics is not fully ergodic and the CV distribution does not come out flat. As mentioned, in the case of well tempered metadynamics the CV distribution is not expected to be flat, but the maximal constrained entropy method is a powerful method to “correct” the free energy by adding ex post further information about the system injecting extra information. In our case we use the maximal constrained entropy to introduce in the computation of the free energy the change of number of hydrogen bonds in α-helices in folding↔unfolding processes. In general the maximal constrained entropy method can be used either to improve the estimate of the free energy for a non-converging system (e.g., in a metadynamics simulation the CV distribution is not flat) or to compute the free energy by reintroducing ex post degrees of freedom related to the CV (like the α-helices’ hydrogen bonds in the case of frataxin, see Section 3). This second use of the maximal constrained entropy method is really powerful because allows to have a reliable estimate of the free energy while keeping efficient the simulations by limiting the degrees of freedom of the system.
2.4 Estimating the free energyThe main goal of this work is to compare the change of free energy as a function of the number of hydrogen bonds (s) computed using well tempered metadynamics and maximal constrained entropy, with the results obtained in protein thermal denaturation experiments Yu et al. (1995); Petrosino et al. (2019). Both BPTI and FXN are folded in a structure where one or two α-helices lay over a small β-sheet. The experimental measurement of the free energy difference between folded and unfolded states was obtained by measuring the molar ellipticity at 222 nm, a wavelength where the contribution of α-helix to CD spectra is dominant. Besides acting on the α-helices arrangement, the protein ternary structure can also be perturbed by destroying the intra-molecular hydrogen bonds that stabilize the β-sheet. For a small protein like BPTI (58 residues), we decided to include in the CV all the hydrogen bonds that are formed in the native folded state Parkin et al. (1996). For FXN (121 residues) we took instead as a CV the number of hydrogen bonds occurring in the β-sheet formed by 4 anti-parallel β-strands, which are observed both in 1EKG and 5KZ5 structures Botticelli et al. (2022). This choice in the case of FXN was made to reduce the number of degrees of freedom of the system thus substantially decreasing the time required to sample its phase space. The use of such CV as a way to monitor the structural transitions in the protein was inspired by several previous applications of metadynamics Barducci et al. (2006).
For both proteins and variants, the biasing potential, VG, was obtained at the end of a systematic construction (well tempered metadynamics) in which VG is progressively built by summing over Gaussian functions of the CV. Gaussian functions (possibly scaled by the biasing factor in the case of the well tempered metadynamics) are deposited every 20 ps along the molecular dynamics (MD) simulation time.
The accumulated final biasing potential, VG(ξ), smoothly interpolated by a polynomial of fourth order, was used for the direct computation of the change in the free energy for folded to unfolded states and vice versa. The free energy change defined in well tempered metadynamics is given by Eq. 6:
Fs−Fs0=−T+ΔTΔTVGs−VGs0(9)with s0 a reference state corresponding to a given value of the CV and VG the external biasing potential determined at the end of construction. Equation 9 holds also in the NpT statistical ensemble, when the construction of the bias VG is performed in such statistical ensemble. In this case, the Helmoltz free energy F(s) is replaced by the Gibbs free energy G(s). We call the latter function G free energy, hereafter, for simplicity. The G free energy extracted from well tempered metadynamics simulations (Eq. 9), was then compared with the G free energy obtained with the maximal constrained entropy method.
The accumulated statistics used in the successive maximal constrained entropy application have been obtained by collecting the system configurations along a trajectory where the biasing potential was kept fixed (i.e., not anymore updated). Within the maximal constrained entropy method, the definition of the G free energy (see La Penna et al., 2004) is given by the formula
Gs=⟨H⟩λ−TkBS̄cs,(10)in which G(s) is written as the combined sum of the enthalpy in the NpT ensemble, and the (informational) entropy measured by the maximal cross-entropy. In Eq. 10 H = U + pV is the enthalpy of the simulated system, λ the parameter associated with the constraint, S̄c the maximal cross-entropy change due to the introduction of such a constraint, kB the Boltzmann constant, and T some effective temperature in the stability range of the system under study. The same free energy definition holds for the Helmoltz free energy F when the enthalpy H is replaced by the energy U if one is working in a NVT ensemble.
The maximal cross-entropy in Eq. 10 is described in the following. Given an estimate, P̃γ, of the metastatistic probability, say the one provided by metadynamics, the problem of finding the least-biased expression of the probabilities Pγ, that is, nearer to P̃γ and satisfies the condition
s=⟨ξ⟩=∑γPγξγ,(11)is solved by determining the maximum of the cross-entropy functional Attard (2000); La Penna (2003); La Penna et al. (2004).
ScP,P̃=−∑γPγlnPγP̃γ.(12)under the constraint (Eq. 11). The well-known solution of this variational problem is given by the formulae:
Pγ=P̃γZλexp−λξγ(13)Zλ=∑γP̃γexp−λξγ(14)with the parameter λ the solution of the (highly non-linear) equation:
s=∑γPγξγ=1Zλ∑γP̃γexp−λξγξγ.(15)The quantity exp (−λξγ)/Zλ is called the modulation factor of the metastatistics. Owing to Eq. 15, λ is a function of s. Inserting the solution for Pγ into Eq. 12 one gets for the cross entropy at its maximum:
The average of H (or simply of U in NVT simulations) is obtained using equations like
bλ=⟨B⟩λ=1Zλ∑γP̃γexp−λξγBqγ,(17)with B either H or U and Zλ=∑γP̃γexp(−λξγ). The identification of Sc and T in Eq. 10 with, respectively, thermodynamic state function entropy S and state variable absolute temperature T, is empirical. It must be noticed that changes in thermodynamic T S values are also reflected in the changes of ⟨H⟩λ as a function of λ.
The details to compute the free energy within the maximal constrained entropy method, the direct calculation of ⟨H⟩λ in Eq. 10 as well as the free energy error estimate is the same we used in our previous work where the “non tempered” version of the metadynamics Botticelli et al. (2022) was employed. In this work we concentrate on collecting more accurate averge quantities (well-tempered metadynamics and longer simulations) and on applying the proposed method also to a simpler protein (BPTI). We must note that the direct calculation of ⟨H⟩λ in Eq. 10 includes the effects of the fluctuations of U and V due to the movement of all explicit water molecules and ions included in the atomistic model of the protein environment. The fluctuations of H are huge, while the change of the average of H with s is small. This is a serious issue when using the total enthalpy like in Eq. 10. As it is customary done in these cases, we use an approximate evaluation of ⟨H⟩λ, where H is replaced by the effective mean-field free energy Ū of the protein solute. The advantage of this approximation is that the energy of the system is thermally averaged over the many degrees of freedom of water molecules and ions surrounding the much smaller solute protein aggregate.
A widely used strategy for the evaluation of the effective mean-field energy of the solute protein is the so-called molecular mechanics/Poisson-Boltzmann solvent accessible approximation (MM/PBSA) Simonson et al. (2002). In this approach the mean-field energy for solute-solvent interactions is described as the sum of polar (electrostatic) and non-polar (surface) contributions. For each protein configuration Q one writes
ŪQ=UintraQ+Gsolv,npQ+Gsolv,polQ,(18)where Uintra is the intra-molecular part of the potential energy in the protein force-field, given by
UintraQ=UstrQ+UbendQ+UtorsQ+UvdwQ+UelQ.(19)The various contributions are the stretching (Ustr), bending (Ubend), and torsional (Utors) terms in the potential. Uvdw and Uel are the Lennard–Jones and Coulomb interactions, respectively, computed by summing over all the pairs of atoms of the protein.
The last two terms in Eq. 18 represent solute-solvent contributions to free energy at fixed Q. Mean field energy is the energy as a function of Q once the variables associated to solvent positions and velocities are averaged for the given value of solute positions Q. The averaging is performed at the given thermodynamic state variables p and T used in the simulation of the whole system. Within this mean-field assumption, the solute and the solvent are made independent. This is a strong approximation, since the chosen collective variable contains the number of hydrogen bonds within protein groups and once a single intramolecular hydrogen bond is broken there is a large chance for the formation of hydrogen bonds with the water molecules in the protein environment where the breaking event occurs. On the other hand, this elementary change of free energy, that does not imply a wide change in protein structure, can be calculated within the MM/PBSA approximation as the sum of Gsolv,np and Gsolv,pol. Therefore, under this approximation, the change of free energy G(s) depends on the number of protein configurations for which a unitary change of s is allowed independently of the configuration of the protein environment. The calculation of Gsolv,np and Gsolv,pol is described in the following.
The term Gsolv,np is the contribution to the solute-solvent free energy due to the formation of a cavity of zero charge density with the shape of the solute protein and the creation of the solute-solvent interface. Introducing a charge density in the space occupied by the solute leads to the Gsolv,pol contribution. The charge density is given in terms of the point charges qi of the atom i sitting at the point r⃗i, where i runs over the Na atoms of the solute molecule.
The term Gsolv,np is calculated as an empirical linear combination of the solvent accessible surface area (SASA) for each group in the solute molecule Ooi et al. (1987) according to the formula
Gsolv,np=∑iNaσiSASAi,(20)where the coefficients σi are positive or negative for hydrophobic or hydrophilic groups, respectively (see below for details). Finally the electrostatic contribution to the solute-solvent free energy, Gsolv,pol, is given by the electrostatic energy required to charge the low-dielectric solute molecule of generic shape into a high-dielectric medium like a salt-water solution. The magnitude of this contribution is obtained by a numerical finite difference solution of the Poisson–Boltzmann equation Rocchia et al. (2002).
2.5 Summary of the methodWe summarize the complicated computational protocol of our theoretical analysis as follows. One starts by performing MD simulations at T = 300 K in the presence of the biasing potential VG(ξ) built according to the well tempered metadynamics strategy. The resulting statistics is what we call metastatistics. Using the set of collected configurations, we determine the λ parameter that maximizes the cross-entropy Sc in the maximal constrained entropy method, under the constraint ⟨ξ⟩ = s. In the case of BPTI, ξ is the number of hydrogen bonds holding together the protein α-helices and β-sheet secondary motifs. In this case, the ξ of metadynamics and that of maximal constrained entropy coincide. In the case of BPTI, differently from FXN (see below), the ξ collective variable takes into account the whole of the secondary structure as it is observed in the crystal folded structure. Therefore, s takes integer values in the range between 0 and 16. In the case of FXN, the variable ξ used in metadynamics is the number of hydrogen bonds holding together the protein β-sheet (made of 4 anti-parallel β strands). The values of s are in the range between 0 and 15. But in the maximal constrained entropy approach we extended ξ adding to it the number of hydrogen bonds in the two α-helices. For each value of s, we get a value of λ that yields the modulating weight
wqt=1Zλexp−λξqt,(21)with q the system configuration at time t, indexing the microstate γ, along the collected metadynamics trajectory. For details see Ref. (Botticelli et al., 2022).
2.6 Simulation parametersApart from the fact that differently than what was done in Ref. (Botticelli et al., 2022), in this work the metastatistics is obtained as altruistic multiple-walkers well-tempered metadynamics, most of the technical details of the simulation procedure we followed to compute the expectation values of the physical quantities of interest described in Section 2 are identical to those reported for FXN in Ref. (Botticelli et al., 2022). Below we only outline the few differences.
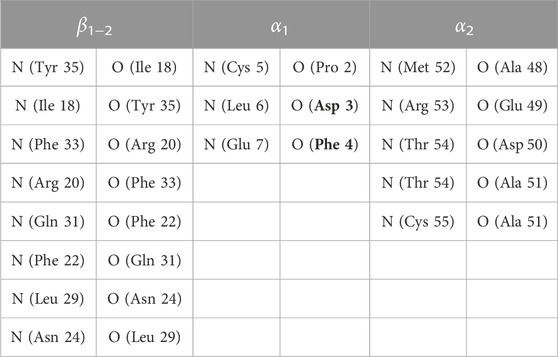
Table 1 provides the list of hydrogen bonds used to define the CV for the BPTI. All the hydrogen bonds contribute to the BPTI CV and are used both in well tempered metadynamics and maximal constrained entropy. For FXN only the number of hydrogen bonds in the β-sheet, β1−4, is used to generate the statistics of metadynamics. However, the total number of hydrogen bonds listed in Table 1 of Ref. (Botticelli et al., 2022), including the two α-helices added to the definition of CV in the successive maximal constrained entropy step. We call this an extension of the CV ξ used in metadynamics and we indicate it with ξ′. The corresponding constrained average is indicated with s′.
TABLE 1. Pairs of atoms used in Eqs 16–18 of Ref. (Botticelli et al., 2022) and related label in parameter S. As for FXN, see Table 1 of the same publication. Residues are those of BPTI WT sequence. Mutated residues are boldface.
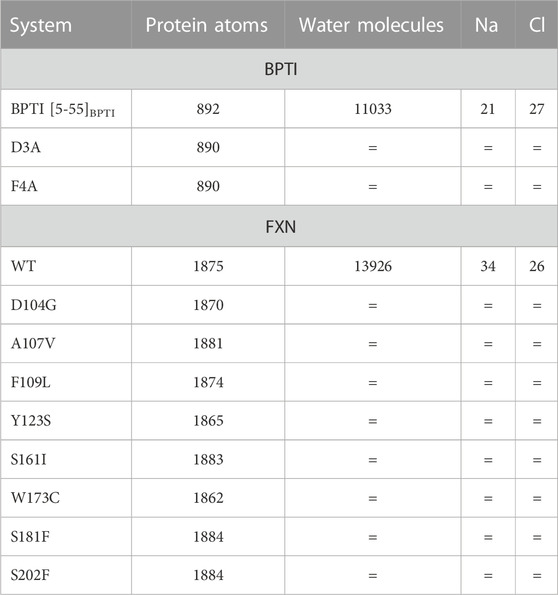
Table 2 reports the number of atoms of the two systems (BPTI and FXN) we have studied. In the case of BPTI, the structure of the unique folded structure available [1BPI PDB entry Parkin et al. (1996)] has been used. As for FXN, the initial configurations of the various walkers are obtained using the available crystallographic information about the native FXN protein sequence. We used two structures: the X-ray structure of the mature human frataxin [PDB 1EKG, segment 88-210 Dhe-Paganon et al. (2000)]; the structure of FXN in the mitochondrial iron-sulfur cluster assembly machine as it was determined by electron microscopy (PDB 5KZ5, chain A, segment 42-210 Gakh et al. (2016)).
TABLE 2. Short description of the atomistic models used in metadynamics simulations. The composition of each system changes only in the protein sequence for each protein (BPTI and FXN, respectively). The number of water molecules and counterions (NaCl) is the same for all the 90 walkers representing each system, and the same (= symbol) for different variants of the same protein.
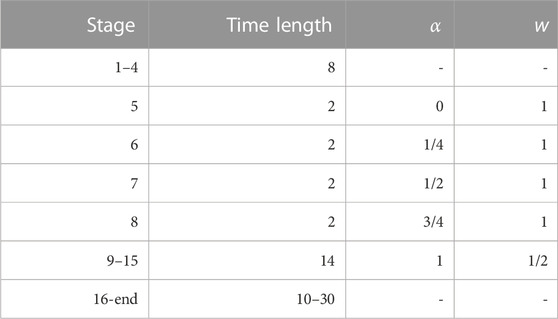
The values of α and w of Eq. 3 in Ref. (Hošek et al., 2017) and used in the successive stages of the simulation are reported in Table 3. As for the construction of the biasing potential, we remark that its construction in the present work lasted 22 ns, while in our previous application it lasted 16 ns. The exchange of the bias among walkers takes place every 2 ns. At the end of stage 15 (see Table 3), i.e., after simulating each walker for a total of 30 ns using an altruistically updated bias, the external bias that will be used in stage 16 and in the following steps is not updated any more. From stage 16 to the end the final metastatistics is collected, storing configurations along the simulated trajectory every ps. The time duration of this last simulation step was 10 and 30 ns for BPTI and FXN, respectively.
TABLE 3. Short description of the simulation stages used to build the external bias VG(ξ) and to acquire the metastatistics at constant external bias. Where α and w are not indicated, the external bias is not updated. The initial bias is zero. Therefore, stages 1–3 (6 ns) are equilibration stages. The bias construction is the same for all variants of BPTI and FXN. As for the constant bias simulation stage 16–20 (10 ns) were collected for BPTI, while 16-30 (30 ns) were collected for FXN. Values of α and w are used when applying the altruistic combination of single-walker updating (2 ns) of VG using Eq. 3 of Ref. (Hošek et al., 2017). The resulting global altruistic bias is used in the following 2-ns stage (next line). The bias after stage 15 is approximately the same for all walkers and, therefore, is made identical for all walkers by averaging over the 90 walkers.
3 Results3.1 Bovine pancreatic trypsin inhibitor (BPTI)48 single point mutations have been studied in the case of BPTI in the literature Yu et al. (1995) via alanine-scanning. This set of mutations includes all residues, with the exception of 6 Ala and 4 Cys, mutated to Ala. The reference sequence used to study the change in melting temperature is the native sequence where Cys 14, 30, 38, and 51 are mutated in Ala. This reference variant is indicated as [5-55]BPTI, to underline the presence of the residual 5–55 disulfide bridge. The sequence is used because the native sequence has 3 disulfide bridges in the folded structure and it does not unfold at T < 100°C. The removal of 2 disulfide bridges allows the melting at T < 50°C, while the protein keeps the same folded structure as the native (WT) sequence, as summarized in Ref. (Yu et al., 1995). Therefore, we could use the structure determined for the WT sequence as the initial representation of the folded state (1BPI Parkin et al. (1996) in PDB).
According to our conventions, a positive ΔΔG means a larger reversible work required to unfold the given variant with respect to the reference sequence. All the variants analyzed in experiments have been already studied as part of large data-sets in previous works dedicated to predictions of free energy change Guerois et al. (2002); Steinbrecher et al. (2017). In our work we are interested in predicting the sign of the free energy change, which is also the sign of the change of the melting temperature Tm, ΔTm. As paradigmatic cases we focused, among the 48 variants, on the two displaying the largest measurable change in the absolute value of ΔTm. The mutations with the most positive and negative value of ΔTm [see Table 1 in Ref. (Yu et al., 1995)] are D3A and F4A, respectively.
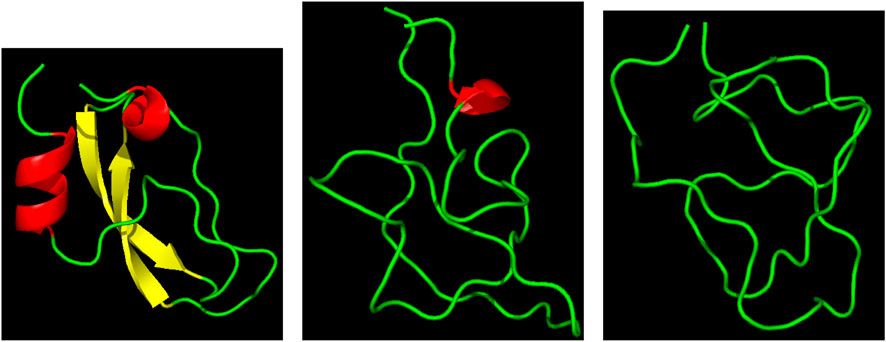
Three representative structures of the [5-55]BPTI reference sequence of BPTI. are displayed in Figure 1 to show how the folded (left panel) and unfolded (right panel) states look like in terms of atomic configurations. Native BPTI is folded into a ternary structure with two short α-helices and a small β-sheet. The construction of the external bias, VG(ξ), perturbs the ternary structure by breaking the intramolecular hydrogen bonds.
FIGURE 1. Three representative structures of [5-55]BPTI reference sequence of BPTI. Left—ξ = 13 (folded state); middle—ξ = 4 (unfolded state); right—ξ = 4 (unfolded state). α-helices are in red; β-sheet is in yellow; the displayed ribbon interpolates the backbone atoms. The Pymol program is used for the molecular drawing Schrödinger (2015).
This is why we decided to take as a collective variable ξ the sum of the number of hydrogen bonds between the two α-helices (α) and the β-sheet (β). The number of hydrogen bonds of α-helices and β-sheets in the initially folded structure (PDB 1BPI) is 8 for both secondary structures. Therefore, the values of ξ span the range between 0 and 16. Figure 1 shows in the right panel that the unfolded state is represented by a molten globule. This occurs because of the short-range nature of the collective variable we have chosen. In the specific case of BPTI the presence of the residual disulfide bridge 5-55 that seals the N-terminus with the C-terminus also pushes the protein towards this atomic arrangement.
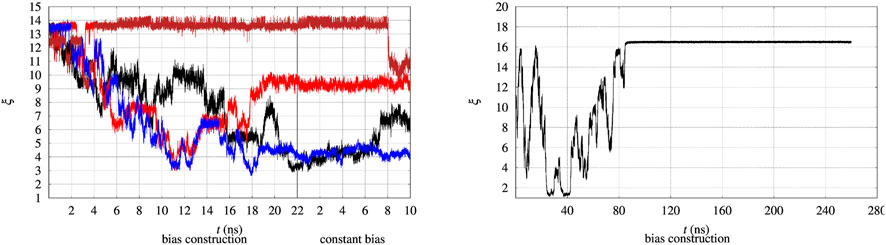
The evolution in time of the collective variable ξ is notoriously slow, even by using well tempered metadynamics. Therefore the convergence of the external bias VG(ξ) is expected to occur after very long simulation times. This issue is illustrated in Figure 2, where the time evolution of ξ of 4 walkers among 90 is displayed. We remind that every 2 ns the bias VG obtained by the whole set of 90 walkers is exchanged among all of the walkers during bias construction in the altruistic approach [Eq. 3 in Hošek et al. (2017)]. Furthermore, before the bias construction the 90 walkers have been separately equilibrated for 8 ns. The figure is divided in two parts. The time evolution during the 22 ns of bias construction is displayed on the lefthand side. The time evolution at constant bias, which constitutes the metastatistics used to compute the biased equilibrium averages, during the last 10 ns is displayed in the righthand side. The figure clearly shows that the unfolding of the protein often occurs during bias construction, since ξ decreases from the value characterizing the folded state to values of 3–4 in 3 cases over the 4 displayed. In certain cases (red curve) the expected behaviour of a random walk of ξ in the 3–14 range is observed.
FIGURE 2. Time evolution of the collective variable ξ during the bias construction (left part) and at constant bias (right part), with the vertical line dividing the bias construction from the bias application. Left—The evolution is displayed in different colors for 4 representative walkers among 90 and for BPTI in the [5-55]BPTI sequence. Right—The same evolution is displayed for a single walker in well-tempered metadynamics of FXN in the WT sequence.
In principle metadynamics is capable of letting the system, starting from the known folded configuration, to unfold and refold. In practice this rarely occurs in affordable computational time, unless the system is sufficiently small. To illustrate the time-scale required for collecting such trajectory, the behaviour of ξ for the longer FXN chain is displayed in Figure 2 (right panel) for a single walker. In this trajectory a single-walker well-tempered metadynamics is performed for 260 ns. While the first 100 ns of the trajectory displays an ideal behaviour for metadynamics [see for instance Figure 2 in Ref. (Barducci et al., 2006)], when the bias is no more effectively updated by new Gaussian functions the system becomes frozen in a fixed configuration. This effect is expected in well-tempered metadynamics, since the height of the Gaussian functions that are added to the bias is progressively decreased by construction.
Anyway, the dynamics of ξ shows that in order to observe a proper random walk of ξ for all walkers, simulation time should have been at least 10–100 times larger. The dynamics of ξ becomes even slower when the bias is kept constant compared to bias construction (righthand side of both panels in Figure 2). This behaviour is due to the effect of noise during bias construction, occurring when new Gaussian contributions ar
留言 (0)