
記住我

The trial will be carried out in at least seven pancreatic cancer centres. Most of the centres are part of the Clinical Trial Network of the German Society of Surgery (CHIR-Net; www.chir-net.de). A list of the trial sites can be found in Additional file 1.
Eligibility criteriaThe following are the preoperative inclusion criteria for patients:
Patients with suspected or histologically verified resectable, borderline or locally advanced pancreatic cancer of the pancreatic head (i.e. pancreatic ductal adenocarcinoma, intraductal papillary mucinous neoplasm (IPMN)-carcinoma or periampullary cancer of the pancreatobiliary-type)
Patients scheduled for elective partial pancreatoduodenectomy (irrespective of neoadjuvant therapy)
Assumed resectability in accordance with the surgical protocol for experimental and control intervention as judged by the treating surgeon
Ability of the subject to understand character and individual consequences of the clinical trial
Written informed consent
Age ≥ 18 years
The following are the intraoperative inclusion criteria for patients (prior to randomisation):
No distant metastases
No paraaortic lymph node metastases
Intraoperative confirmation that the patient can be operated on according to both surgical methods (experimental or control group)
The following are the exclusion criteria for patients:
Participation in another interventional trial with the interference of intervention and outcome of this trial
American Society of Anesthesiologists (ASA) grade > 3
Distant metastatic disease
Eligibility criteria for trial centresAll participating trial sites will be high-volume centres with broad expertise in pancreatic surgery and have the necessary expertise, equipment and personnel to perform this trial.
Who will take informed consent?All patients scheduled for PD will be screened preoperatively with regard to the inclusion and exclusion criteria. An authorised investigator will inform the patient, orally and written, about the aims of the trial, the possible risks, the procedures, the possible hazards to which he/she will be exposed and the mechanism of treatment allocation (randomisation). The written informed consent form will be signed and personally dated by the patient according to the ICH guidelines in Good Clinical Practice.
Additional consent provisions for collection and use of participant data and biological specimensNo additional biological samples will be collected during the trial.
InterventionsExplanation for the choice of comparatorsAs outlined in the introduction, radical extended PD including the removal of the tissue in the TRIANGLE area could improve the R0 resection rate and thereby the survival of patients suffering from pancreatic cancer. So far, high-quality data in terms of a multicentre randomised controlled trial on the TRIANGLE intervention are lacking. Therefore, a trial evaluating the effect of the TRIANGLE intervention compared to standard PD according to the current guidelines is needed.
Intervention description Exploration phase (experimental and control group)The experimental intervention is limited to the resection phase. Thus, both groups (experimental and control) start with the same exploratory phase. After safe access to the abdominal cavity, the exploration phase should comprise the following steps, which can be performed in any order (at the discretion of the surgeon):
1.Exclusion of hepatic metastases and peritoneal carcinomatosis. If distant metastases are present, the patient must not be randomised.
2.Dissection of the gastrocolic ligament to open the lesser sack and define tumour extent along the pancreatic gland and the stomach. No transection is performed at this time.
3.Any type of artery-first approach may be performed to explore potential tumour infiltration [20]. If resection of the tumour is not possible based on intraoperative findings, the patient must not be randomised.
4.A Kocher manoeuvre is performed to expose the para-aortic lymph nodes (LNs) and the root of the SMA.
(a)If a suspicious para-aortic LN is detected, it should be resected and sent for frozen section.
(b)In case the frozen section procedure reveals tumour infiltration, no randomisation should be performed as available data shows that the prognosis of patients with positive para-aortic LNs is poor (ISGPS guidelines [21]).
5.After the exclusion of distant and paraaortic LN metastases and intraoperative confirmation that the patient can be operated according to both experimental or control groups, randomisation will be performed.
Resection phase—experimental group (TRIANGLE operation)The experimental intervention consists of two parts:
ADissection of the SMA according to level 3 described by Inoue et al. [13]. This step includes a dissection of the nerve plexus around the superior mesenteric artery (plSMA) from at least 5 to 11 o’clock (180°). A wider resection (≥ 180°) up to a circular (360°) resection of the lymph plexus and plSMA is allowed and at the discretion of the surgeon. Performing this step should result in a circular (360°) dissection of the superior mesenteric vein (SMV).
BComplete dissection of the soft tissue in the “triangle” between CA, SMA and MPA [14].
Resection phase—control groupPatients randomised to the control group will receive standard PD with dissection of the SMA according to Inoue level 1 or 2 and standard lymphadenectomy according to the German S3 guidelines [8] but no TRIANGLE operation. This constitutes the current standard of care.
Deviations from the described control intervention with venous or arterial reconstruction or extended pancreatectomy in case of advanced tumour infiltration are possible as long as there is no radicality necessary according to Inoue level 3 along the SMA or removal of soft tissue in the triangle area.
Resection phase—experimental and control groupsThe further key steps of the resection phase in the experimental as well as the control group should include lymphadenectomy in the hepatoduodenal ligament as described in the German S3 guidelines [8]. Furthermore, the transected end of the bile duct and the transection of the pancreatic body should be sent for frozen section to rule out microscopic tumour infiltration. In case of tumour infiltration in the pancreatic body, further resection of the pancreatic body is indicated. Transection of the postpyloric duodenum or the prepyloric stomach should be performed as oncologically necessary. Therefore, pylorus-preserving, pylorus-resecting or classical Whipple procedures or any variants are allowed in the trial. If extended pancreatic resections (e.g. total pancreatectomy) or resection of neighbouring organs are necessary, patients remain in the trial. Details should be given during visit 2 (surgery). Any kind of venous or arterial resection and reconstruction can be performed as deemed necessary by the operating surgeon.
Reconstruction phase—experimental and control groupsThe reconstruction phase is the same in both groups including pancreatojejunostomy or pancreaticogastrostomy, hepaticojejunostomy and a duodeno- or gastrojejunostomy according to local standards. Similarly, the placement of drains, abdominal wall and skin closure should be performed according to local standards. Details will be recorded during visit 2.
If reconstruction needs to be performed later in a second operation, this is possible and does not result in the exclusion of the patient from the trial.
Criteria for discontinuing or modifying allocated interventionsAfter inclusion into the trial, patients still have to meet the intraoperative inclusion criteria to be finally randomised to one of the intervention groups. If during the exploration phase distant metastases or paraaortic lymph node metastases are found or it can not be confirmed that the patient can be operated on according to both surgical methods (experimental or control group), the patient has to be excluded intraoperatively and will be defined as an intraoperative drop-out. Furthermore, if, in the investigator’s opinion, continuation of the trial intervention would be detrimental to the subject’s well-being, the investigator may stop the trial intervention for this patient. In this case, the reason for the individual premature trial termination must be recorded in the electronic case report form (eCRF) and in the patient’s medical records.
Strategies to improve adherence to interventionsAs the interventions of the TRIANGLE trial (experimental and control groups) are surgical interventions, strategies to improve adherence to the allocated intervention are not needed.
Relevant concomitant care permitted or prohibited during the trialAdditional perioperative care of the patients will be performed according to the institutional standards. All additional medications and/or treatments are permitted during the trial when considered necessary by the treating physician.
Provisions for post-trial careThe post-trial care of the patients will be the regular tumour follow-up according to the current guidelines. No compensation will be provided.
Outcomes Primary endpointThe primary endpoint of the TRIANGLE trial will be the DFS after resection, defined “as the time from randomisation until disease recurrence or death from any cause” [22]. Disease recurrence can be a local recurrence or distant metastases. Given the overall dismal prognosis of PDAC, the high rate of recurrence as well as the objective of the trial, i.e. to reduce recurrence rates and improve OS, DFS seems the logical and most relevant endpoint for the TRIANGLE trial. It is a frequent endpoint in cancer trials in the adjuvant setting after definitive surgery, and it is widely accepted by regulatory authorities worldwide [22, 23] and has been used in previous randomised controlled trials (RCTs), thus allowing the comparison of data between trials [16, 24, 25]. Furthermore, it is based on objective and quantitative assessments. DFS is patient-relevant as it includes all-cause mortality and recurrence. In order to avoid detection bias, follow-up will be standardised for both groups and include clinical outpatient visits, contrast-enhanced CT scan and tumour markers every 6 months for 3 years.
Primary estimandIn the recently released addendum to the ICH E9 guideline (final version), the estimand framework is recommended as clear and transparent definition of “what needs to be estimated to address a specific scientific question of interest”. Such an estimand should be defined through the treatment condition of interest, the population of interest, variable of interest, specification of how intercurrent events are handled, and summary measure. The specification of how intercurrent events are handled is referred to as intervention effect in the following. This way a more precise definition of the treatment effect of interest in relation to the trial objective(s) is enabled. Based on such an estimand, adequate methods to estimate this estimand can be chosen. In the following, the primary estimand (see Table 1) corresponding to the primary objective is described.
Table 1 Overview of the primary estimandTreatment: The treatments patients will receive in the experimental group or in the control group of the trial are specified as mentioned above.
Population: The targeted population is defined through the in- and exclusion criteria.
Variable: The variable is disease-free survival after resection, defined as the time from randomisation until disease recurrence (local recurrence or distant metastases) or death from any cause.
Intervention effect: Possible intercurrent events and the strategies to handle them are as follows: Death, as an intercurrent event occurring after randomisation, is handled by inclusion into the definition of the primary endpoint which reflects a composite strategy. Patients with incomplete observation time due to loss to follow-up or early drop-out will be censored at the last observation, which reflects a hypothetical strategy. Besides these events, other post-randomisation events (e.g. intervention not as randomised, discontinuation of chemotherapy) will not be considered, thus reflecting a treatment policy approach, which means that the effect of randomised treatment is estimated irrespectively of other post-randomisation events not captured in the primary endpoint definition. This corresponds to the intention-to-treat (ITT) principle.
Summary measure: The summary measure is the adjusted hazard ratio.
Secondary endpointsSecondary endpoints of the TRIANGLE trial are as follows:
1.Rate of the following:
(a)Microscopically complete margin clearance (> 0.1 cm margin clearance, R0(CRM-))
(b)Microscopic margin clearance ≤ 0.1 cm (R0(CRM+))
(c)Microscopic margin involvement (R1) resections according to the 8th edition of the UICC TNM classification
2.Rate of the following PD-associated postoperative complications within 90 days after the index operation:
(a)Postoperative pancreatic fistula (POPF) as defined by the International Study Group of Pancreatic Surgery (ISGPS) [26]
(b)Postpancreatectomy haemorrhage (PPH) as defined by the ISGPS [27]
(c)Delayed gastric emptying (DGE) as defined by the ISGPS [28]
(d)Bile leakage as defined by the International Study Group of Liver Surgery (ISGLS) [29]
(e)Lymphatic fistula as defined by the ISGPS [30]
(f)Diarrhoea as graded by the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 [31]
3.All other postoperative complications graded according to the Dindo-Clavien classification [32] within 90 days. This endpoint complements the PD-associated complications evaluated via the above-mentioned ISGPS endpoints, to assess all remaining postoperative complications to establish a risk-benefit assessment of the two interventions.
4.Overall survival within the study period.
5.Local recurrence within the study period.
6.Quality of life (QoL) according to the European Organisation for Research and Treatment of Cancer (EORTC) QLQ-C30 and PAN26 at discharge and during every follow-up visit compared to baseline.
7.Quality of recovery (QoR) according to the QoR-15 questionnaire on postoperative day 5 compared to baseline.
8.Length of primary hospital stay in days from the day of index operation to the day of discharge.
9.Serious adverse events (SAEs) in both groups .
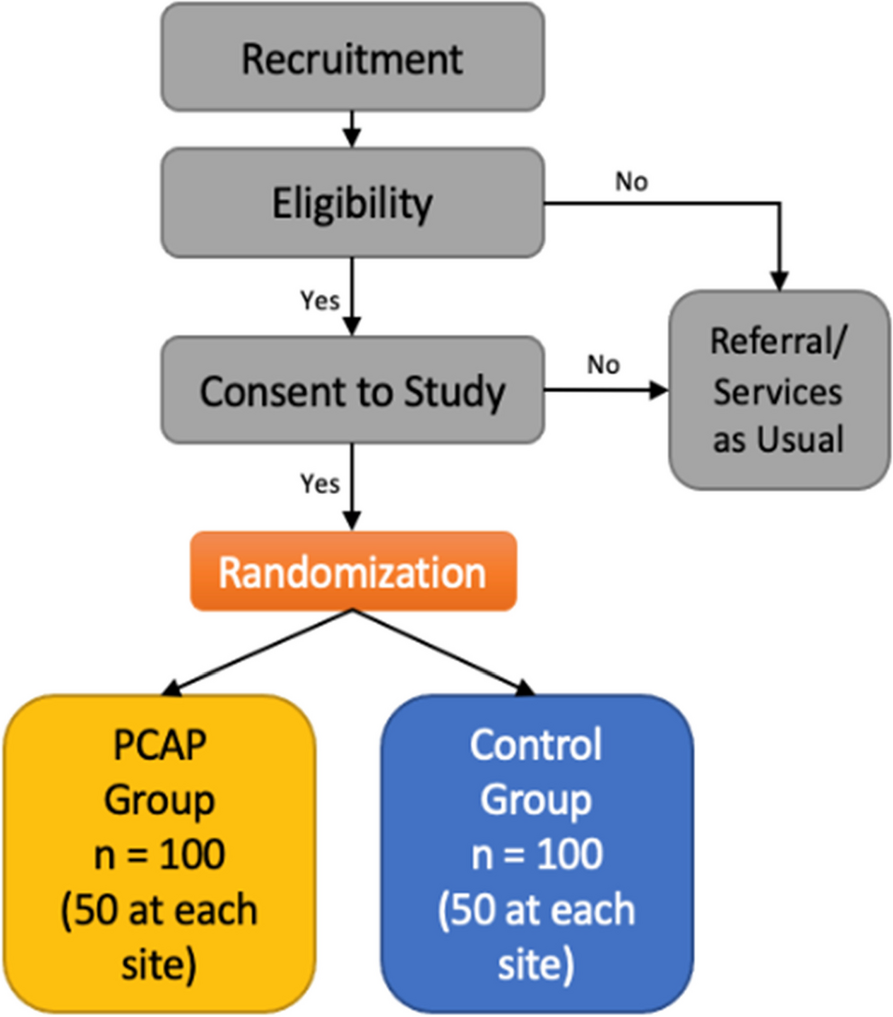
Participant timelinePatients scheduled for elective PD will be screened preoperatively (visit 1). After the patient has given informed consent, the inclusion and exclusion criteria are assessed during the screening visit. Patients fulfilling all the inclusion criteria and none of the exclusion criteria and who consent to take part in the trial are randomised during surgery (visit 2; surgery and randomisation) after the surgeon has obtained certainty that both interventions can be performed in the patient. The results of the pathologic work-up (secondary endpoint) will also be recorded in the eCRF section of visit 2. Patients are planned for follow-up visits on postoperative days 5, 10–12 and 90 (visits 3, 4 and 6) for the evaluation of primary and secondary endpoints. Visit 5 will be performed at discharge. If discharge occurs before visit 3 or 4, the respective postoperative visits (3 or 4) can be omitted. In addition, 6, 12, 18, 24, 30 and 36 months (visits 7–12) after surgery, patients are planned for follow-up visits to evaluate primary and secondary outcome parameters (Fig. 1 and Table 2).
Fig. 1
Flow chart of the TRIANGLE trial. F/U, follow-up; POD, postoperative day; PD, partial pancreaticoduodenectomy; R, randomisation. *Respective visits are skipped if the patient has been discharged before
Table 2 Trial visits and documented parametersSample sizeThe sample size calculation is based on the primary endpoint disease-free survival. The assumptions are based on a median survival time of 14 months in the control group and 21 months in the treatment group resulting in a corresponding hazard ratio of 0.667 assuming exponentially distributed survival times [7, 16, 33]. To detect the assumed difference using a log-rank test at a significance level of 5% (two-sided) with a power of 80%, a total number of 192 events (i.e. recurrence or deaths) are required for the entire trial. It is expected that applying the Cox proportional hazards model, as described below, will lead to an increase in power. With an accrual period of 30 months and a follow-up period of at least 36 months, and assuming an exponentially distributed drop-out rate of 20% (at 66 months), n = 270 (135 per group) patients are needed (based on the approximation formula by Schoenfeld). The sample size calculation was done using ADDPLAN 6.1.1.
RecruitmentIn order to recruit the necessary number of patients, at least seven pancreatic cancer centres will participate in this trial. All participating trial sites will be high-volume centres with broad expertise in pancreatic surgery and have the necessary expertise, equipment and personnel to perform this trial. Most of the centres are part of the CHIR-Net.
Assignment of interventions: allocationSequence generationTo ensure an equal distribution of patient characteristics and confounders between the two groups, a randomisation tool will be used. Randomisation will be performed at the patient level with a centralised online randomisation system (www.randomizer.at). The online randomisation procedure provides information regarding the group allocation and a randomisation number. The randomisation sequence is computer-generated and will be stratified by centre and presence/absence of neoadjuvant treatment. Block randomisation will be performed. The randomisation procedure will be performed intraoperatively (after surgical exploration) after the fulfilment of all intraoperative inclusion criteria.
Concealment mechanismAs randomisation in the TRIANGLE trial depends on the intraoperative inclusion criteria and the findings during the exploratory phase of surgery, the allocation to the assigned group will be performed intraoperatively after fulfilling the inclusion criteria. Allocation concealment is therefore not necessary, as the assigned intervention will be performed immediately after randomisation.
ImplementationThe randomisation will be performed intraoperatively by a member of the study team after fulfilling all of the intraoperative inclusion criteria by using the online randomisation tool. Names of the randomising study team member as well as the team of surgeons will be documented. They will not be involved in the outcome assessment.
Assignment of interventions: blindingWho will be blindedPatients and outcome assessors will be blinded to the intervention in order to guarantee an unbiased assessment of the primary and secondary endpoints. The outcome assessors will be neither part of the surgical team that performs the trial intervention nor have access to the randomisation tool. All patient-reported outcomes (PROs) will be assessed using validated measures. Moreover, the discharge letter will contain no information regarding group allocation.
Procedure for unblinding if neededIf unblinding is necessary due to medical conditions, it can be performed by the treating physicians or one of the unblinded study team members. Unblinding will be documented and reported to the steering group of the trial.
Data collection and managementPlans for assessment and collection of outcomesAll of the included trial sites will be high-volume centres with broad expertise in pancreatic surgery as well as the conduction of clinical trials. Most of the centres are part of the CHIR-Net, which has successfully performed trials with similar indications and recruitment rates in the past [34,35,36].
To further enhance data quality and to minimise detection bias, validated measures and classifications will be used where possible (see the “Outcomes ” section). Additionally, the primary outcome (DFS) is an objective measure defined “as the time from randomisation until disease recurrence or death from any cause” according to international guidelines [22].
Plans to promote participant retention and complete follow-upAs outlined above, all of the included trial sites will be experienced in the conduction of randomised trials with similar indications and follow-up procedures. Furthermore, pancreatic cancer patients are in need of an oncological follow-up that usually is performed in high-volume centres. If follow-up is not performed in the centres, visits after discharge of the patients can also be done by telephone including a collection of the necessary data.
Data managementAn eCRF will be used for data collection. The study data will be collected using REDCap [37] (Research Electronic Data Capture), a secure, web-based data capture application hosted at the IMBI. To assure a safe and secure environment for the data acquired, data transmission is encrypted with secure socket layer (SSL) technology. The database server is located in a secure data centre and is protected by a firewall. The system provides an infrastructure to support user roles and rights. Only authorised users are able to enter or edit data; the access is restricted to data of the patients in the respective centre. All changes to data are logged with a computerised timestamp in an audit trial. All clinical data will be pseudonymised. Backups are conducted regularly.
All protocol-required information collected during the trial must be entered by the investigator or designated representative in the eCRF. For health-related quality of life and patient-reported outcome data, patients may directly enter the data in the eCRF. Alternatively, paper-based reported outcome questionnaires must be entered by the investigator or designated representative in the eCRF. The investigator or designated representative should complete the eCRF forms as soon as possible after information is collected, preferably on the same day that a trial subject is seen for an examination, treatment, or any other trial procedure. Any outstanding entries must be completed immediately after the final examination. An explanation should be given for all missing data. The completed eCRF must be reviewed and signed by the local investigator or by a designated sub-investigator.
To guarantee high data quality, data validation rules will be defined in a data validation plan. Completeness, validity and plausibility of data will be checked at the time of data entry (edit checks) and using validating programs, which will generate queries. The investigator or the designated representatives are obliged to clarify or explain the edit checks and queries. If no further corrections are to be made in the database, eCRF data will be locked.
All data collected will be integrated into a statistical analysis system. During study conduct, database access will be granted to the data manager only. After database closure, access rights will be granted to the biometricians as well. The data will be managed and analysed in accordance with the appropriate standard operating procedures (SOPs) valid in the IMBI Heidelberg that guarantee an efficient conduct complying with Good Clinical Practice (GCP). Photo files from the surgery are collected at each local site and are stored according to the applicable local, national and international regulations.
ConfidentialityPatients will be informed as to the strict confidentiality of their data, but that their medical records may be reviewed for trial purposes by authorised individuals (trial monitor) other than their treating physician. It is the responsibility of the investigator to maintain patients’ confidentiality. During the trial, patients will be identified solely by means of their individual identification codes. Trial-specific documents will be stored in accordance with local data protection law/ICH-GCP Guidelines and will be handled in the strictest confidence. For the protection of these data, organisational procedures are implemented to prevent the distribution of data to unauthorised persons. The patients’ data will be transferred in a pseudonymised form from the trial centre to cooperating partners (coordinating investigator, data management). Names and all confidential data of participating patients will be handled in line with the obligations of medical secrecy, the European General Data Protection Regulation (Datenschutzgrundverordnung, DSGVO), the Federal Data Protection Act (Bundesdatenschutzgesetz) and the state Data Protection Act (Landesdatenschutzgesetz). Participating patients’ data will be documented in the eCRF only in pseudonymised form. Decoding of the pseudonymised data is only permitted in justified cases. Third parties have no access to original documents. After completion of the trial, data collected during the study will be kept on file for 10 years. It is guaranteed that the data protection provisions are followed. The sponsor provides data protection management and an information security management system. The Clinical Trial Centres are contractually obliged to comply with the DSGVO and other data protection regulations.
Plans for collection, laboratory evaluation and storage of biological specimens for genetic or molecular analysis in this trial/future useThere are no plans for the collection, laboratory evaluation and storage of biological specimens for genetic or molecular analysis in the TRIANGLE trial.
留言 (0)