
記住我


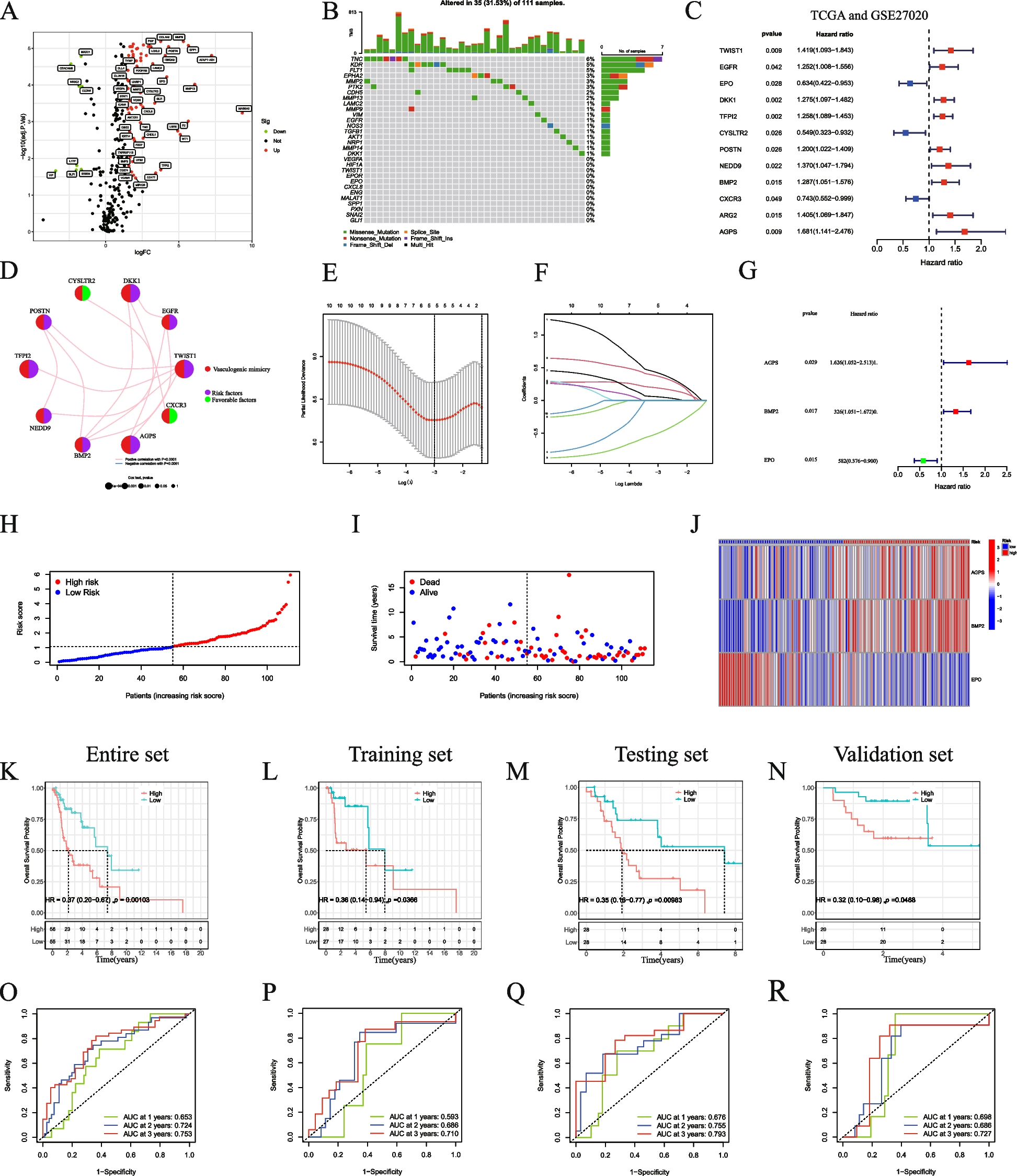
Diapedesis is a process by which leukocytes pass through the vascular endothelium into the sub-endothelial space. It may occur in the paracellular or transcellular forms, known as inter- and intracellular ways, and includes cellular adhesion, movement, and migration [1]. Many studies reported the roles of some genes in the paracellular and transcellular ways of diapedesis. The PECAM1 and CAV1 gene families are reported to transfer leukocytes through the vessel wall [2] so that the downregulation of PECAM1 causes to the accumulation of leukocytes within the basement membrane [3, 4]. Some genes are also reported in the leukocyte trafficking pathway [2, 5, 6]. These genes may be regulated by non-coding RNAs, including miRNAs [7]. The adhesion molecules through their receptors, in addition to being involved in cell–cell interactions, can transduce the bidirectional signals between the endothelial cells and leukocytes causing to vascular permeability [8,9,10,11,12]. Moreover, chemokines through the CXCR1 and CXCR2 receptors activate the integrins and adhesion molecules via G and beta-arrestin proteins [13, 14]. Duffy antigen receptor for chemokine (DARC), known for binding to CXC family, is responsible for delivering the chemokines by the endothelial cells. Furthermore, IL6 causes to express the adhesion molecules and some chemokines, such as CCL2 through GP130 in neutrophils [14]. The blood cells also migrate via other receptor/ligand complexes during diapedesis process. The N-formyl-methionyl-leucyl-phenylalanine (FMLP) binds to formyl-peptide receptor (FPR) and causes cell migration [14]. The expression of CD99 on endothelial cells is required for neutrophil trans-endothelial migration [15]. Furthermore, the neutrophils and macrophages are involved in the inflammatory responses via FC (free cholesterol) receptor [10, 16]. Adenosine also increases neutrophil chemotaxis and phagocytosis, albeit at low concentrations, through the adenosine receptor subtypes [17]. RAP1, RAP2 and Rho Family can regulate endothelial permeability and leukocyte trans-endothelial migration [18]. The cellular permeability also increases by TNF-α through tight junctions (TJ), adherent junctions (AJ) and actin filaments. It is also increased by the histamine and bradykinin through VE-cadherin-related pathways [18]. Adhesive leukocyte signals through GEF-small Rho GTPase axis quickly affect cell–cell connections. The effect of Rho, together with radial stress fiber, leads to the increased vascular permeability. The Rho-cross talked pathways also mediate actomyosin contraction led to cellular migration [12]. Furthermore, the P-Rex/Rac signaling pathway increases vascular permeability by producing ROS compounds [12]. The CD99 also facilitates the movement of leukocytes through TRPC6 calcium channels and adhesion molecules such as ICAM1, VCAM1 and PECAM1 on the endothelial cells [11, 19, 20].
The leukocytes are the first line of blood cells in vascular permeability via the chemokine-followed inflammatory events [8, 21,22,23]. Some diseases associated with chronic inflammation, such as lupus and psoriasis are highly exposed to cardiovascular diseases [24]. When the inflammation begins, in the first stage, endothelial cells absorb leukocytes [8] so that the cellular rolling occurs by adhesive reactions. During adhesion, cellular morphology is changed: first round, then flat, and finally rounded [8]. Furthermore, the recruitment of leukocytes by activating memory T cells is followed by inflammatory cytokines [23]. Moreover, chemical adsorbents when attach to their ligands, lead to the movement of leukocytes, a process that is called chemotaxis [25, 26]. The complementary components C3a and C5a can be involved in absorbing inflammatory cells [26, 27]. Other factors such as N-formyl peptides [28], VWF, and WPB [29] may cause vascular inflammation. It is well known that during inflammation, APO-A1 changes in HDL particles so that its decrease is associated with an increase in VWF [29]. Also, cellular connections on endothelial cells change the permeability in response to a series of compounds. For example, a set of pro-inflammatory stimuli such as thrombin and histamine increase permeability while sphingosine 1-phosphate and angiopoietin-1 which act as anti-inflammatory agents, reduce cellular permeability [18]. In vitro, the FMLF bacterial peptide binds to FPRs and causes neutrophil migration and polarization [9, 30]. Adenosine is also known an inflammatory modulator, so the adenosynthetic system is suggested as a therapeutic target [31].
As indicated in the above, the neutrophils, leukocytes, and T cells migrate through activated arterial walls, which is a prerequisite step due to the infection or damage [32]. The entry of cells into the inflamed locations occurs in several stages including the weak adhesion, rolling, and crawling between endothelial cells and leukocytes [8, 33] (Fig. 1). The leukocyte adhesion cascade is sequentially performed by selectin and integrin on the cell surfaces [32]. In addition to the endothelium, leukocytes must pass through the pericyte layer, the basement membrane of blood vessels. The neutrophils, however, do not pass through pericyte bodies but through pericyte gaps [32]. In this way, IL1β increases the expression of several genes, including VCAM1, CX3CL1, MCP1 and IL6 in pericytes [34]. Furthermore, the CXCL8 secretion from pericytes by IL1β, LPS and TNF-α progressed the transport of neutrophils [32]. Moreover, pericytes express MHC2 when stimulated with cytokines, which increase phagocytosis capability of neutrophils [32]. The polar shape that cells take on is necessary to cross the endothelial barrier. The areas where leukocytes leave the vessel, are called low expression regions (LERs), have low extracellular matrix proteins such as laminin 8, laminin 10, collagen IV and nidogen [32]. The strong adhesion is mediated by a set of adhesion molecules in the immunoglobulin superfamily, including leukocyte integrins that bind to endothelial ligands such as ICAM1 and VCAM1. β2 Integrin is also essential for the transcellular movement of leukocytes. LFA1 and MAC1 are expressed by neutrophils and cause to adhere tightly and control cell crawling [32, 33]. It is also well known that during migration, actin filaments are responsible for the polymerization of the main edge of the cell, but actomyosin prevents protrusion in the lateral membrane [8]. When leukocytes begin the transmigration process, the appearance of the leukocytes is rounder than their predecessor (crawling) just before they leave. Glycocalyx, at the apical surface of endothelial cells, immobilizes chemokines to promote integrin-induced adhesion [21]. Platelets may be one of the modulators of inflammatory reactions in atherosclerosis via binding to endothelium by integrin and ICAM1 [35]. As indicated in these studies, the endothelial function is impaired, the diapedesis is disrupted and blood cell outflow occurs excessively in the atherosclerosis process. These blood cells including monocytes can be polarized into macrophages and develop the atherosclerosis process in vessel sub-endothelial space.
Fig. 1
Leukocyte paracellular and transcellular diapedesis ways. The roles of adhesion molecules on Capture (1), Rolling (2), Adhesion (3) and Crawling (4) of leukocytes. Some genes involved in transcellular (5.A) and paracellular (5.B) ways. BioRender.com
Macrophage polarization affects the atherosclerosis processThe monocytes polarize to different macrophages in vessel sub-endothelial space. Some agents involved in the macrophage polarization, characteristics and their roles in the development of atherosclerosis are explained in the following. Atherosclerotic plaques contain mostly M1 and M2 macrophages. The microenvironment surrounding macrophages can acquire different phenotypes. For example, interferon γ and LPS activate the M1 macrophage [36]. M0 macrophages can be converted to M1 and M2 macrophages by LPS/IFN-γ and IL4/IL-13 [37]. The subgroups of M2 macrophages are known as M2b, M2c, M2a and M2d [38,39,40]. M0 is polarized by IL4/IL13, LPS/IL1β and, IL10/TGF-β for the generations of M2a, M2b and, M2c macrophages, respectively [41]. M0 macrophage is also polarized into M3 macrophages through TGF [42] (Fig. 2). M4 macrophages feature both M1 and M2 macrophages, and are induced by CXCL4 [38]. In inflammatory conditions, macrophages form M1, while polarization to M2 occurs under anti-inflammatory conditions [43]. M1 macrophages are abundant in areas prone to shoulder rupture, but M2 macrophages are found in the areas such as adventitia [36, 44]. M4 macrophages express proinflammatory chemokines such as TNF-α, IL6, MMP7 and MMP12. Furthermore, M4 macrophages have HLA–DR less than M1/M2 macrophages [45] (Fig. 3).
Fig. 2
Polarization of monocyte to M3 macrophage. BioRender.com
Fig. 3
Polarization of monocyte to M4 macrophage. BioRender.com
In atherosclerotic lesions, the macrophages scavenge ox-LDL particles and can convert into foam cells. In this state, the macrophages produce large amounts of ROS so that these compounds decrease M2 macrophage polarization [46, 47]. The polarization of macrophages towards M2 by mir-27a also occurred [7]. The atherosclerotic plaque stability is associated to calcification and the kind of polarized macrophages in ruptured areas. The M2 macrophage is reported to eliminate inflammation while the M1 is involved in the progression of plaque, so it is proposed that the M2 macrophages play an important role in the prevention of inflammation by secreting IL-10 [36]. M1 macrophages are seen in primary atherosclerotic lesions, while M2 macrophages are more in advanced lesions. M4 macrophages can also be seen in atherosclerotic lesions and cause the instability of fibrous cap in the plaque [45].
The polarization of macrophages towards M1 macrophage is done by Th1 (T helper 1), TNF-α and GM-CSF [48]. However, M1 macrophages have low IL10 values, but IL6 and IL1β produced by these macrophages are responsible for the progression of inflammation [48]. Furthermore, TLR ligands (LPS) and other cytokines, such as IFN-γ are involved in the formation of M1 macrophages. CXCL4 cytokine leads to the polarization of monocytes to M4 macrophages [40, 45]. By treating M1 macrophages with IL4, or using IL13, the polarization can be converted to reveal anti-inflammatory phenotypes [49]. When damage occurs, M1 macrophages begin to produce a variety of inflammatory molecules such as TNF-α, inducible nitric oxidase synthase (iNOS) and IL-12 [49]. LncRNA-COX2 is contributed to the elevation of iNOS, IL12 and TNF-α, which are more pronounced in M1 macrophages [40]. M1 and M2 macrophages have different arginine metabolism; M1 macrophages, primarily through iNOS use arginine to produce nitric oxide, while M2 macrophages convert arginine to ornithine and urea through arginase [50]. M2 macrophages can induce macroscopic calcium deposition by VSMC maturation and osteoblastic differentiation, called macrocalcification, which is associated with chronic inflammation [36]. M2 macrophages have low levels of IL12 and IL23, however, they have high IL10 values. IL10, M-CSF and Th-2 cytokines stimulate the generation of M2 macrophages [49, 50]. M2 macrophages produce anti-inflammatory cytokines including IL4, IL5, IL10, IL13 and TGF-β [48, 49]. STAT1 mediates M1 macrophage activation, while STAT6 mediates M2 macrophage activation [49]. STAT1 and STAT6 can inhibit each other; that is, when STAT6 is activated, it suppresses STAT1-dependent transcription and vice versa [49]. IL4 can activate STAT6 and the cell phenotype moves towards M2 [28, 47]. However, STAT3 induces M2c Macrophage by IL-10 [40]. MCP-1 (monocyte chemotactic protein-1) is also reported as influential factors on macrophage polarization.[7] M2b markers include IL10, CCL1, LIGHT, CD86, SPHK1, TNF-α, and IL6. To identify M2b, however, the IL10 marker is not suitable and other markers should be checked because all these M2 macrophages secrete IL10 abundantly. LncRNA GAS5 suppresses the CCL1 gene. The CCL1 is essential for the polarity of M2b macrophages [40]. Also, miR-223 is considered to contribute for M2 polarization [40]. Both M1 and M2b macrophages contain CD86, so this marker cannot distinguish M1 from M2b macrophages. However, it is useful to recognize M2b from other subclasses of M2 macrophages. TNF-α is also secreted by both M1 and M2b macrophages [40].
The macrophage polarization relates mainly to the activation of some pathways such as PI3K/AKT, NF-KB, STAT1/STAT6 and MAPK/ERK [50]. Adiponectin decreases the expression of some cytokines via the inhibition of NF-KB so that it directs M2 macrophages via AMPK and PPARα pathways. Adiponectin may also increase the secretion of other cytokines, such as TNF-α, IL6, and IL12. However, there were some controversies on the role of adiponectin as a pro-inflammatory or anti-inflammatory factor [50]. MAPKs are involved in the polarization of M2b macrophages through the activation of ERK1/2, p38, and JNK. MBL, mannose-binding lectin, inhibits the signaling pathways related to MAPK and NF-KB, resulting in the decrease of polarity of M2b macrophages. Moreover, the polarization of M2b macrophages can be done by PI3k pathway [40] (Fig. 4). On the other hand, the polarization of M1 macrophages can be done by NOD-Like receptor related pathways [51]. The P65 and P50 subunits of NF-κB can create pro-inflammatory and anti-inflammatory phenotypes in macrophages. When the NF-κB P65 subunit is activated, it promotes the polarization of M1 macrophages (Fig. 5). The M2b macrophage is polarized by the activation of P50 subunit. NF-κB and IRF are activated by ALD-DNA (activated lymphocyte-derived DNA), which are involved in the polarization of macrophages towards M2b phenotype [40]. M1 phenotype also occurs with INF-γ, which acts through the STAT1 pathway. The PI3K/AKT signaling pathway is activated by IL4 and its effect on the formation of macrophage phenotypes depends on its isoforms. The P110γ isoform causes the M1while the P110αβγ isoform makes M2 phenotype. AKT2 and AKT1 pathways are also involved in the M1 and M2 phenotypes, respectively. It also reported that the macrophage anti-inflammatory phenotype produced by IL4 is involved with MAPK/ERK signaling pathway [50].
Fig. 4
Polarization of monocyte to M2 macrophage. Several signaling pathways contribute to polarize the different forms of M2 macrophage such as M2a, M2b and M2c. BioRender.com
Fig. 5
Polarization of monocyte to M1 macrophage. BioRender.com
The macrophage population is originated from different cells and maintained by different agents in tissues. The conventional theory that each tissue macrophage originated from the bone marrow circulatory monocytes is changed by reporting that macrophages from embryonic progenitors may persist into maturity and self-maintain. In a few cases, tissue-resident macrophages are completely embryo-derived, including microglia inside the brain, while others are continuously substituted from monocytes (LP) [
留言 (0)