
記住我



The dried Citrus (1000 g) was extracted three times with 75% ethanol for 3 h (3000 mL × 3) under reflux. The filtrate was concentrated under decompression. The collected mass was dissolved in distilled water (300 mL), then adjusted to a pH of 2 with HCl (20%, g/g) and washed with ethyl acetate (300 mL × 3) to remove acidic compositions. The rest water part was adjusted pH to 9 using ammonia water (23%, g/g), and separated with ethyl acetate (300 mL × 3). The combined ethyl acetate was dried with anhydrous sodium sulfate and evaporated under decompression to generate CAE (1.8% yield). Some components of CAE—N-Methyltyramine, Synephrine, Flavanone, Hesperitin, Limonin, Narirutin, Hesperidin, Tangeretin and Sinensetin—and other ingredients were identified by LC-MS (4600 UPLC/Triple TOF) and shown in Additional file 1.
A murine model of LPS-induced pulmonary fibrosisMale C57BL/6 mice are kept at room temperature (25 ± 1 °C), atmospheric humidity (50% ± 10%), under a regular 12-hour dark light cycle, and fed with standard laboratory food and water. Animal welfare and experimental procedures were carried out strictly in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, the United States) and the related ethical regulations of our university. Animal studies were in compliance with the ARRIVE guidelines [15].
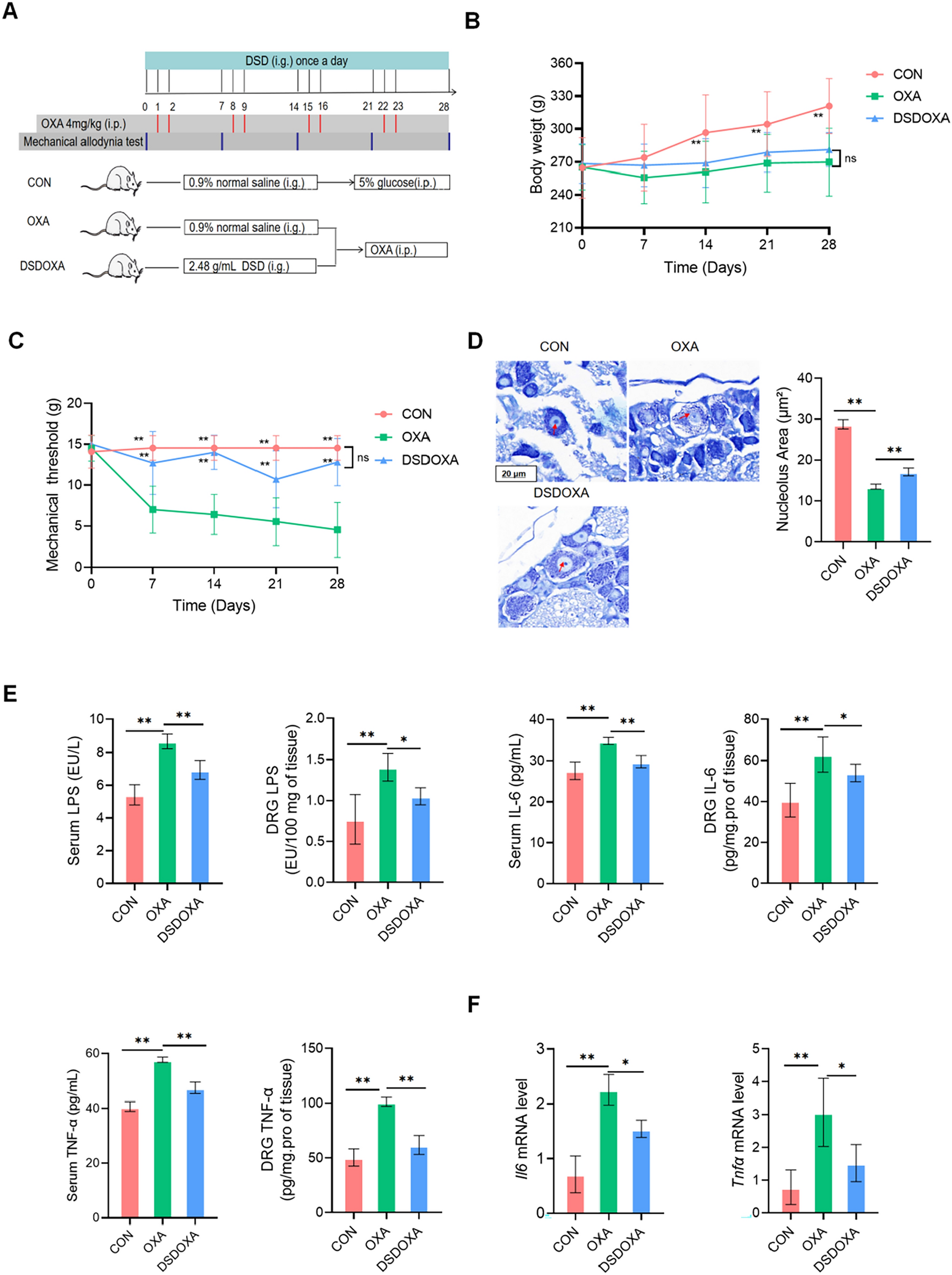
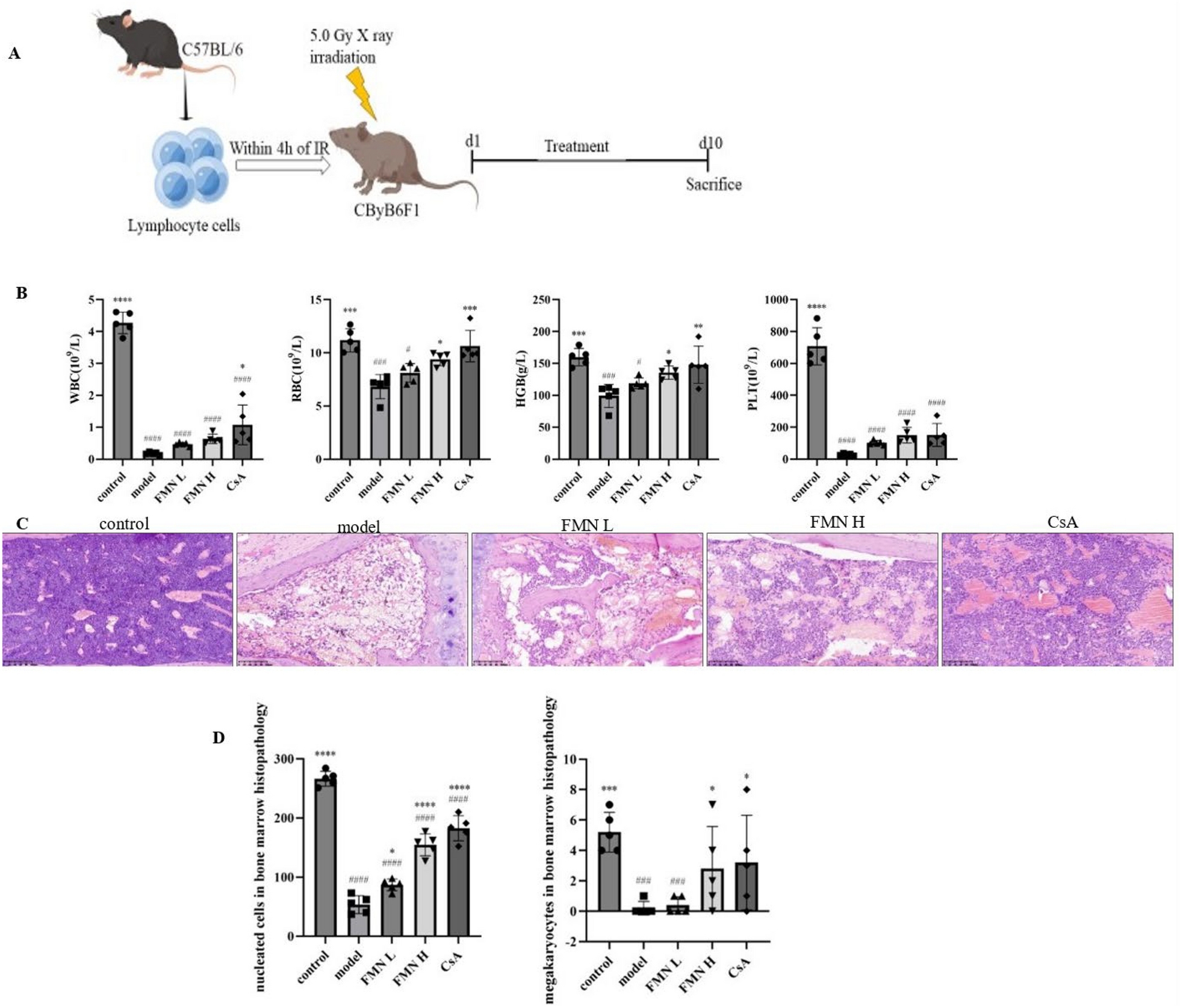
The pulmonary fibrosis model was established by intraperitoneal injection of purified LPS extracted from the membrane of Escherichia coli 0111:B4 (Sigma-Aldrich, St. Louis, MO, USA) at 5 mg kg− 1 day− 1 in a total volume of 50 µl from the 1st to the 5th day [11]. The preliminary experiment revealed that 5-day-injected LPS caused pulmonary fibrosis on the 14th day (data not shown), and then we chose the 14th day as a separation. CAE administration was categorized into pretreated groups (gavaged from the 1st to the 14th day, and shown as Pre) and therapeutic groups (gavaged from the 15th to the 28th day). Dexamethasone (Dex) was used as positive control. Details are as follow: Ninety-six of 8-week-old C57BL/6 male mice weighing 18–22 g were randomly divided into 12 groups: Alone (normal saline), LPS (5 mg kg−1 day−1), Pre Control (0.5% CMC-Na for the former 14 days), LPS + Pre H-CAE (96 mg kg−1 day−1, pretreated), LPS + Pre M-CAE (64 mg kg−1 day−1, pretreated), LPS + Pre L-CAE group (32 mg kg−1 day−1, pretreated), LPS + Pre Dex (5 mg kg−1 day−1, pretreated), Control (0.5% CMC-Na for the later 14 days), LPS + H-CAE (96 mg kg−1 day−1), LPS + M-CAE (64 mg kg−1 day−1), LPS + L-CAE group (32 mg kg−1 day−1), LPS + Dex (5 mg kg−1 day−1). And after 28 days, all of the mice were sacrificed. The method of CAE administration was shown in Fig. 1A and B. Part of the lung tissue was fixed with 10% formalin to prepare paraffin, and other parts were quick-frozen with liquid nitrogen and stored at −80 °C for later use. Dex was purchased from Sigma-Aldrich (St. Louis, MO, USA). And CAE was extracted, separated and identified in laboratory as described previously [16].
Fig. 1
CAE ameliorated LPS- induced lung injury and pulmonary fibrosis. A, B Time chart of mouse model building. A Administration procedure of the pretreated groups. B Administration procedure of the therapeutic groups. C, D Mice body weight change. E H&E staining. F Masson staining. G Sirius Red staining. The scale bar is 100 μm. H, I Area density analysis of Masson’s trichrome and Sirius Red staining, respectively. Results were shown as the means ± SEM (n = 8). *P< 0.05, **P < 0.01, ***P < 0.001 vs. the LPS group, # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Alone group
Tissue sectioning and histopathologyLung specimens fixed in 10% buffered formalin and were embedded in paraffin blocks. Paraffin sections of tissues were placed on glass slides, and paraffin was removed with xylene. The slices were handed over to Service B io Technology Co., Ltd (Wuhan, China) for serial staining include Hematoxylin and Eosin, Masson’s Trichrome and Sirius Red staining to evaluate the severity of fibrosis. Examinations were performed and photographs were captured with a light microscope. ImageJ software (National Institutes of Health, Maryland, USA) was used to calculate the ratio of the area with positive expression to the total field of Masson’s Trichrome and Sirius Red staining.
Immunohistochemistry stainingLung specimens fixed in 10% buffered formalin and were embedded in paraffin blocks. Paraffin-embedded lung sections were heat-fixed, deparaffinized, rehydrated, antigen retrieval, blocked with 3% goat serum and incubated with anti-α-SMA antibody (1:200, Cell Signaling Technology, MA) or anti-COL1A1 antibody (1:200, Cell Signaling Technology, MA) or anti-E-cadherin antibody (1:150, Cell Signaling Technology, MA) or anti-N-cadherin antibody (1:150, Cell Signaling Technology, MA) overnight at 4 °C, then the slides were detected using Real Envision Detection kit (GeneTech, Shanghai, China) according to the manufacturer’s instructions. Observe the sections with a microscope and take pictures. Image J software calculated the ratio of positive expression area to the total field of immunohistochemical staining of α-SMA, COL1A1, E-cadherin and N-cadherin.
Measurement of collagen I and hydroxyproline contentsThe contents of collagen I and hydroxyproline in lung tissues were determined by commercially available kits obtained from Nanjing Jiancheng Bioengineering Inc. (Catalog #H589, Catalog #A030-2-1). Hydrolysate (1 mL) was added to serum (500 µL) or lung tissue (50 mg, wet weight) and incubated in water at 95 °C for 20 min for hydrolysis. The sample solution was adjusted to a pH of 6.0 to 6.8 using the pH modulation A and B solution in the kit. Double distilled water was added to 10 mL and took 3 mL to add 20 mg of carbon, then centrifuged and retained the supernatant. The detection solution sequentially added in the enzyme plate according to the operation table of the instructions and incubated in water at 60 °C for 15 min and centrifuged after cooling. The supernatant was determined the absorbance at a wavelength of 550 nm.
Cell cultureA549 (human lung adenocarcinoma) cell line was obtained from the China Center for Type Culture Collection (Wuhan, China) and was cultured in 25-cm2 flasks (Corning, Corning, NY, USA) containing Dulbecco’s modified Eagle medium (DMEM; Gibco, Thermo Fisher Scientific, Waltham, MA), containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Waltham, MA) and 1% penicillin streptomycin (Biological Industries, Haemek, IsCAEl), at 5% CO2 and 37 °C. To create the in vitro fibrosis stress model, A549 cells were treated with TGF-β1 (10 ng·mL−1, Pepro Tech, USA) for 48 h while which they were treated with three different doses of CAE (250 µg mL−1, 500 µg mL−1 and 1000 µg mL−1) or sivelestat sodium (100 µg mL−1, Catalog #C105530, Chemegen, USA) [17, 18] .
CCK-8 assayCells were seeded in 96-well plate and incubated with various doses of CAE for 48 h. And then added 20 µL CCK-8 Antibody Blocking Peptide (Bioss, Beijing, China) to each well, placed the culture plate in the incubator for 4 h. Finally, the absorbance at 450 nm were measured with a microplate reader. The survival rate of cells was calculated based on the absorbance value of each well.
Western blot analysisA549 cells were seeded in a 6-well plate, the treatment groups were given different doses of CAE (250 µg mL−1, 500 µg mL−1 and 1000 µg mL−1) and TGF-β1 (10 ng mL−1), while the model group was given TGF-β1 (10 ng mL−1) and the control group was given the same volume of DMSO as the CAE after cell adherence. After stimulating and culturing for 48 h, cells were collected and lysed using RIPA Lysis Buffer (Beyotime, Shanghai, China) and quantitated by BCA Protein Assay Kit (Beyotime, Shanghai, China), obtained protein lysates were degenerated at 100 ℃ for 5 min and separated by 10% SDS-PAGE and electrophoretically transferred onto poiyvinylidene fluoride (PVDF) membranes (Millipore Corp, Bedford, MA). The membranes were blocked with 3% BSA (BioFroxx, Guangzhou, China) for 1 h at room temperature, then incubated with specific primary antibodies including anti-E-cadherin antibody (1:1000, Cell Signaling Technology, MA) or anti-N-cadherin antibody (1:1000, Cell Signaling Technology, MA) or anti-α-SMA antibody (1:1000, Cell Signaling Technology, MA) or anti-p-stat3 antibody (1:1000, Cell Signaling Technology, MA) or anti-p-stat6 antibody (1:1000, Cell Signaling Technology, MA) or anti-β-catenin antibody (1:1000, Cell Signaling Technology, MA) or anti-Wnt-1 antibody (1:1000, Santa Cruz Biotechnology) or anti-COX2 antibody (1:1000, Abcam, CA) overnight at 4 ℃, and then incubated with a horseradish peroxidase (HRP) coupled secondary antibody. Protein bands were visualized using Western blotting detection system according to the manufacturer’s instructions. Image J software calculated the gray-scale value of the bands.
CHIP assayCrosslinking DNA and proteins in A549 cells using 1% formaldehyde solution under physiological conditions. Chromatin was broken down by ultrasound, followed by the addition of anti-EZH2 antibody for positive control, anti-IgG antibody for negative control, or anti-Egr1 antibody precipitated crosslinked complex. DNA fragments bound to antibodies were precipitated. Perform de-crosslinking to purify DNA fragments. DNA sequences specifically bound to antibodies are screened by RT-qPCR. The reverse (5′GGAAATGGCTCTGGACTTGGCGGTA3′) and forward (5′GGAGGCAGCCGTTCGGAGGATTATT3′) pair of primers was used to amplify PTEN promoter. The reverse (5′TTGGTGGTTGGGTGATGGAG3′) and forward (5′GATTGCAAGCCCCAATCCC3′) pair of primers was used to amplify COL1A1 promoter.
Immunofluorescence stainingA549 cells were cultured at 24-well plate and TGF-β1 (10 ng mL−1) was added in a medium of cells with CAE (500 µg mL−1, 1000 µg mL−1) for 48 h. Then the medium was aspirated and the cells were gently shaken with PBS buffer. The cells were fixed in 1% paraformaldehyde for 30 min and were permeabilized with 0.1% Triton X-100 in PBS for 20 min at room temperature. Nonspecific binding sites were blocked by incubating the cells with 3% goat serum for 1 h. The fixed cells were incubated with a primary antibody against N-cadherin (1:200, Cell Signaling Technology, MA) or E-cadherin (1:200, Cell Signaling Technology, MA) overnight at 4 °C, followed by incubation with fluorescence (FITC)-conjugated goat anti-rabbit IgG (1:1000, Thermo Fisher Scientific, USA) for 1 h at room temperature. The cellular nuclei were stained with DAPI for 15 min. All samples were imaged with a light microscope. Image J software (NIH) was used to calculate the ratio of the area with positive expression to the total field of N-cadherin and E-cadherin.
Immunoprecipitation studiesA549 cells were cultured at 10 cm dish and treated with 1000 µg mL−1 CAE in the presence of 10 ng mL−1 TGF-β1, while the model group was given TGF-β1 and the control group was added same volume of DMSO as the CAE. Then the culture dishes were placed in an incubator for 48 h (with 5% CO2 and 37 °C). A small part of the harvested cell lysates was used for western blot, and the rest of the lysates was incubated with 4 µg GSK3β antibody (1:1000, Proteintech, USA) at 4 °C overnight and precipitated with Protein A/G Magnetic Beads (Thermo Fisher Scientific, USA) for another 1 h at room temperature. The beads were washed 4 times with washing buffer (PBS buffer with 0.05‱ Tween 20) on the magnetic stand and the immunoprecipitated proteins were boiled for 10 min in loading buffer. Finally, the immunoprecipitated proteins were detected by western blot.
SOD, MDA and NO assayA549 Cells seeded in 6-well plate and the supernatant of cells were collected respectively after 48 h. Cells were sonicated by ultrasonic disruptor and quantitated by BCA Protein Assay Kit. Superoxide Dismutase (SOD) assay kit (WST-1 method; Nanjing Jiancheng Bioengineering Inc.), Cell Malondialdehyde (MDA) assay kit (Colorimetric method; Nanjing Jiancheng Bioengineering Inc.) and Nitric Oxide (NO) assay kit (Nitrate reductase method; Nanjing Jiancheng Bioengineering Inc.) were used to measure the activity of SOD and the levels of MDA in A549 cells and the levels of NO in culture medium according to the manufacturer’s instructions, which were used to estimate antioxidants and oxidation products to explore the effect of CAE on cell redox reactions.
Quantitative PCRTotal RNA were extracted from A549 cells and reverse transcribed to cDNA and subjected to quantitative PCR, which was performed with the BioRad CFX96 ouchTM Real-Time PCR Detection System (BioRad, CA) and threshold cycle numbers were obtained using BioRad CFX manager software. The program for amplification was 1 cycle of 95 °C for 2 min followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The primer sequences used in this study were 5′- TTCAGTATCACAACCTCAGCAAG-3′ (forward) and 5′- TGGACCTGCAAGTTAAAATCCC-3′ (reverse). The relative amount of iNOS gene was normalized to the amount of β-actin, and then reported as fold change of basal level.
RNA-seq analysisA549 cells were seeded on 10 cm culture dish and treated with 1000 µg mL−1 CAE in the presence of 10 ng mL−1 TGF-β1 (marked as CAE group), while the model group was given TGF-β1 (marked as TGF-β1 group) and the control group was added same volume of DMSO as the CAE. Cells were collected after incubation for 48 h (with 5% CO2 and 37 °C) and total RNA was then isolated using RNAiso Plus (Takara, Beijing, China) according to the manufacturer’s instructions. RNA-seq was performed at the Applied Protein Technology Co., Ltd in Shanghai, China. RNA-seq of each group was repeated with three independent biological replicates.
Network pharmacology analysisThe targets of the principal components of CAE are retrieved on the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) database (https://old.tcmsp-e.com/tcmsp.php). The UniProt ID of the targets were then searched through the Universal Protein Resource (UniProt) database (https://www.uniprot.org), with the species defined as “Homo sapiens.” The targets of EMT were obtained from GeneCards (https://www.genecards.org/). Target intersection was mapped by Venny (https://bioinfogp.cnb.csic.es/tools/venny). The overlapping targets were selected and imported into the STRING database (https://string-db.org) to obtain the protein interaction relationship. The results were then imported into Cytoscape 3.9.1 software to fabricate and anatomize the Protein-Protein Interaction (PPI) Network.
Statistical analysisData are expressed as the Mean ± standard error of mean (SEM). Difference between multiple groups was analyzed by one-way ANOVA with Tukey’s post-hoc tests. Difference was considered to be statistically significant when P-value < 0.05.
留言 (0)