
記住我



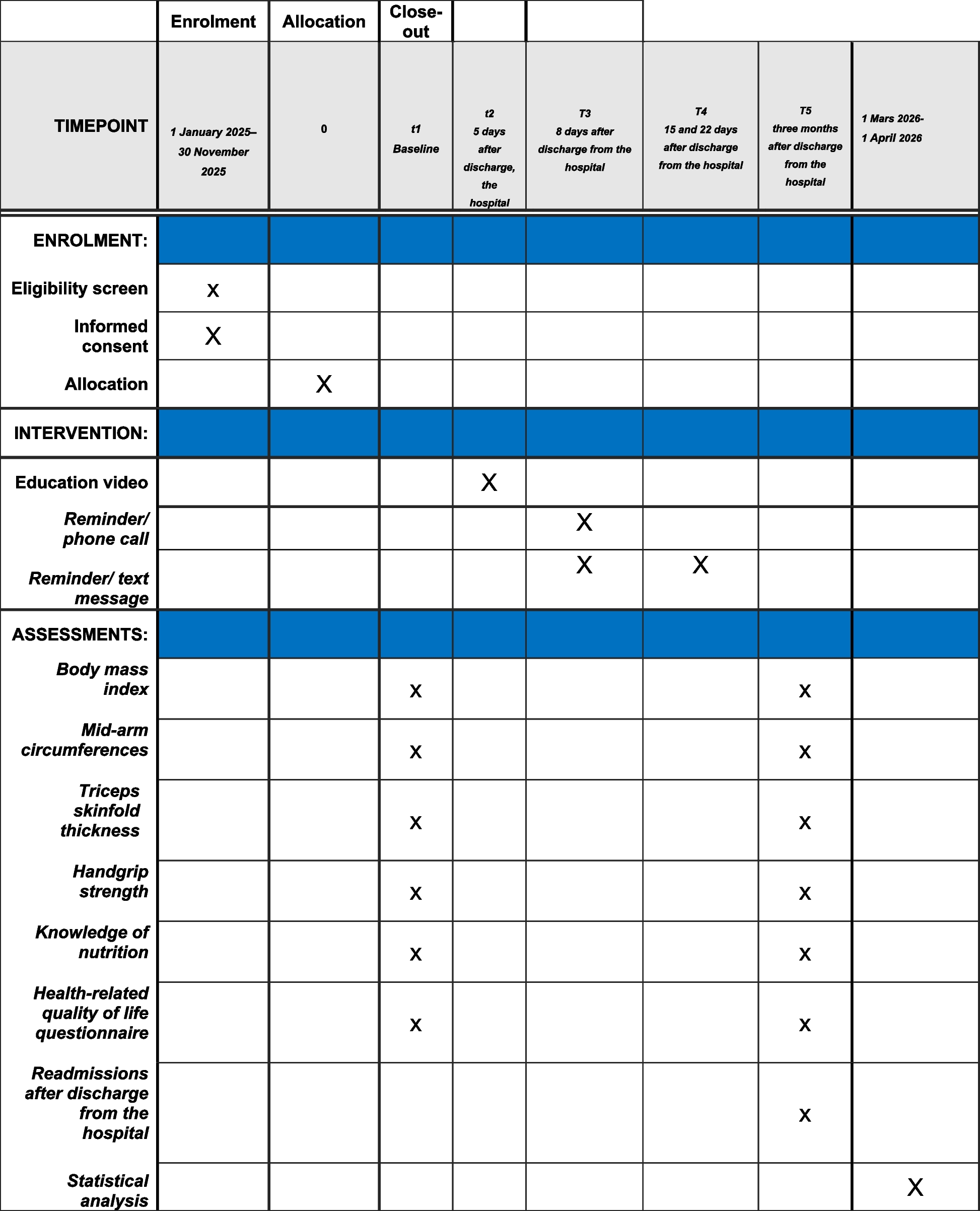
The IB1001-301 trial is conducted in accordance with the International Conference for Harmonisation (of Technical Requirements for Pharmaceuticals for Human Use)—Good Clinical Practice Guideline, the General Data Protection Regulator, and the Declaration of Helsinki. The study protocol was designed in accordance with the SPIRIT 2013 statement and the study is conducted in accordance with the SPIRIT reporting guidelines [15]; the SPIRIT Figure for both the Parent Study and the Extension Phase is included as Supplementary Tables 1 and 2, and the SPIRIT checklist has been completed. The study has been approved by the ethics committees of each participating center and the regulatory authorities in each respective country. The safety, integrity, and feasibility of the trial is monitored by an independent data safety monitoring board (DSMB) consisting of three independent, non-participating members (including two clinicians and a statistician). The function of the DSMB is to monitor the course of the studies and, as applicable, recommend to the sponsor of the trial for discontinuation, modification, or continuation of the study. The roles and responsibilities of the DSMB are defined in a DSMB charter.
Patient population and eligibility criteriaPatients will be screened for eligibility according to the inclusion and exclusion criteria. To be eligible for the respective study, patients aged ≥ 4 years with a confirmed diagnosis of NPC must present with clinical symptoms, provide appropriate informed consent, and undertake a washout of any prohibited medications (if applicable). These include any variant of N-acetyl-DL-leucine (e.g., Tanganil™). For a detailed description of the inclusion and exclusion criteria, see Table 1.
Table 1 Inclusion and exclusion criteria for patient selection in the Parent StudyRecruitment and patient involvementThe principal investigator at each site will be responsible for the enrolment of patients. Patients will be screened at 16 centers across Australia, the Czech Republic, Germany, the Netherlands, Slovakia, Switzerland, the UK, and the USA. The list of study sites is available via www.clinicaltrials.gov (NCT05163288). Patients will be recruited via personal correspondence, routine care appointments, and referrals. In addition, there is collaboration and support from multinational patient organizations representing these rare disease communities. All eligible patients who agree to participate in the study are provided with a full verbal explanation of the trial and the Patient Information Sheet. This includes detailed information about the rationale, design, and personal implications of the study.
Study design and proceduresThe IB1001-301 clinical trial is a double-blind placebo-controlled crossover trial. Patients will be first assessed during a baseline period (with or without a study run-in) and randomized (1:1) to one of two treatment sequences: IB1001 followed by placebo, or vice-versa. Each treatment period will last approximately 12 weeks (84–91 days). Patients will be assessed twice during each period to allow an assessment of intra-patient variability.
ScreeningAt the initial screening visit, patients will be classified as either “naïve” or “non-naïve” depending on their use of prohibited medications within the past 6 weeks (42 days). The schedule of events during the initial screening visit and throughout the baseline period (through visit 1) will vary depending on the patient’s classification as either “naïve” or “non-naïve.” Given the known unlicensed use of the racemate (Tanganil™), for all patients, a urine sample will be taken at visit 1 to detect N-acetyl-D-leucine using a validated liquid chromatography mass spectrometry/mass spectrometry method. Provided the level of N-acetyl-D-leucine is below the permitted threshold, the initial screening visit will be confirmed as visit 1 (baseline 1). If a patient classified as “Naïve” unexpectedly tests positive for levels of N-acetyl-D-leucine above the permitted threshold, at the direction of their principal investigator, a run-in wash-out period of 6 weeks (42 days) is requested before they are eligible to return for a repeat visit 1. Patients who fail two urine N-acetyl-D-leucine tests (e.g., visit 1 and repeat visit 1) are ineligible for the study.
RandomizationAt visit 2, patients will be randomized (1:1) to their respective sequence. Patients will be centrally randomized using Medpace’s ClinTrak Interactive Response Technology (IRT) web-based system. Medpace is responsible for the generation of the allocation sequence, and the IRT system will automatically assign participants to their respective sequence. The study utilizes a permuted block design for randomization. The LIVE randomization list is generated by an independent statistician based on the approved randomization plan. As the study is double-blinded, patients, their families, the study team, and the sponsor will be blinded to the randomization scheme and the sequences to which the patient is assigned. The randomization list will not be available to any person involved in the conduct of the study or the evaluation of the trial until the trial database is locked. The bioanalytical laboratory staff are authorized to receive the randomization list prior to the study conclusion to determine which samples should be analyzed according to standard operating procedures. As the trial is a crossover design, in which each patient will receive IB1001 and placebo, and serve as their own control, no stratification by age is performed.
Intervention periodsFigure 1 displays the naïve and non-naïve study schemes for the Parent Study. Suppl. Tables 1 lists the schedule of enrolment and assessments together with pre-planned time points for clinic visits. Patients who have completed Visit 6 of the Parent Study have the opportunity to continue treatment with N-acetyl-L-leucine (IB1001) in an Extension Phase if the principal investigator determines it is in their best interest. The Extension Phase consists of a 1-year (351–379 day) treatment period followed by a 6 weeks (42–56 days) washout period. Table 2 lists the inclusion criteria for the Extension Phase.
Fig. 1
Parent Study schema. a Naïve patients screening pathway: patients who have not used any prohibited medications within 42 days of screening are “naïve.” Their initial screening visit is treated as visit 1 (baseline 1). b Non-naïve patient screening pathway: patients who have used or are unable to confirm or deny if they have used, any prohibited medication within the past 42 days are “non-naïve.” Patient will be given the opportunity to undergo a minimum of 42 days washout before returning for a repeat visit 1 (baseline 1). From visit 2 onward, the visit schedule is the same for naïve and non-naïve patients
Table 2 Inclusion criteria for patient participation in the Extension StudyFigure 2 displays the Extension Phase study schema. Suppl. Table 2 lists the schedule of enrolment and assessments together with pre-planned time points for clinic visits in the Extension Phase.
Fig. 2
Extension Phase schema. Patients will be assessed approximately 4 times over a 64-week period: at the start of the Extension Phase, after 6 months of treatment, 1 year of treatment, and after a 42-day (+ 14 day) post-extension-phase treatment washout
Study drugThe dosage form of N-acetyl-L-leucine or the matching placebo is granules for oral suspension in a sachet (manufactured by Patheon France S.A.S., Bourgoin Facility, France) which are suspended in 40 mL water, orange juice, or almond milk. A marked measuring cup is provided to each patient.
Administration and study drug dosageDuring the treatment periods for both treatment sequences, the dosing of the study drug is as follows: patients aged ≥ 13 years or aged 4–12 years weighing ≥ 35 kg will take 4 g/day (2 g in the morning, 1 g in the afternoon, and 1 g in the evening). Patients aged 4–12 years weighing 25 to < 35 kg will take 3 g/day (1 g in the morning, 1 g in the afternoon, and 1 g in the evening). Patients aged 4–12 years weighing 15 to < 25 kg will take 2 g/day (1 g in the morning and 1 g in the evening). Medication should be taken at least 30 min before or at least 2 h after a meal. Compliance will be assessed upon a review of the inventory of IB1001 sachets returned by patients.
Study objectivesThe Parent Study and Extension Phase enable the investigation of both the symptomatic (12-week) and long-term (1-year) safety and efficacy of treatment with N-acetyl-L-leucine. The primary objective of the Parent Study is to evaluate the efficacy of N-acetyl-L-leucine versus placebo based on the Scale for the Assessment and Rating of Ataxia (SARA) or modified SARA (mSARA). In the Extension Phase, the primary objective is to evaluate the effects of N-acetyl-L-leucine based on the modified (5-domain) Niemann-Pick disease type C Clinical Severity Scale (NPC-CSS).
For both study phases, the secondary objectives are:
To assess the clinical efficacy (symptomatic and long-term) of N-acetyl-L-Leucine on symptoms of ataxia, functioning, and quality of life for patients with NPC
To evaluate the safety and tolerability of N-acetyl-L-leucine at 4 g/day in patients with NPC aged ≥ 4 years and older
Extension Phase only: characterize the pharmacokinetics of N-acetyl-L-leucine in NPC patients
Safety and efficacy parametersPrimary efficacy endpointThe original SARA scale is an eight-item clinical rating scale (range 0–40, where 0 is the best neurological status and 40 is the worst). It is a reliable and valid clinical scale with a high internal consistency that measures the severity of symptoms and ataxia and increases with disease stage [16].
The original unmodified SARA was selected as the primary endpoint based on advice from EU National Regulatory Authorities (including Germany, Portugal, Spain, and the Netherlands) and the UK Medicines and Healthcare product Regulatory Authority. In the USA, the primary endpoint is the modified SARA (mSARA). This modification was based on the explicit advice of the US Food and Drug Administration, which requested the instrument be modified to:
Include the domains that are the most clinically meaningful towards understanding the functional aspects of ataxia/symptoms in NPC patients
Remove domains where there is no/little movement and which may impact the interpretation and power of the tool (i.e., improve the reliability and sensitivity of the instrument)
Accordingly, the mSARA was developed as a six-item clinical rating scale consisting of the original Gait, Speech Disturbance, Finger Chase, Nose-Finger test, Fast Alternating Hand Movement, and Heel-shin slide domains. It ranges from a score of 0 to 30, where 0 is the best neurological status and 30 is the worst.
The SARA scale will be the basis for the primary efficacy estimand. The mSARA scale will be considered as a supplementary estimand. Withdrawal from study medication due to adverse events and the taking of prohibited medication will be considered as intercurrent events (ICEs) and a treatment policy strategy will be considered for these ICEs. Withdrawal or early termination from the study for unspecified reasons and lost to follow-up will not be considered as ICEs, and the subsequent data will be considered as “missing.” Should these events occur, a last observation carried forward approach will be adopted based on observations at visits 2, 3 for period 1 and visits 4, 5 for period 2.
The model for analysis will be an analysis of covariance model with the difference in the SARA scores at the end of periods I and II as the dependent variable and SARA at baseline (visit 2) as the independent variable together with an indicator variable for sequence [17]. The estimated coefficient of the indicator for sequence will provide the least squares estimate of the difference in the treatment means on division by 2. Secondary endpoints will measure other symptoms and evaluate quality of life (Spinocerebellar Ataxia Functional Index (SCAFI) [18]; Modified Disability Rating Scale (mDRS) [19, 20]; Quality of Life EQ-5D-5L for patients aged ≥ 18; EQ-5D-Y for patients aged < 18 years [21]; and Investigator, Caregiver, Patient Clinical Global Impression Scale (CGI)) [22]. Descriptive statistics will be provided for these measures at each visit, and select secondary endpoints will be evaluated statistically based on a comparison of the period differences (period II–period I) between the two treatment sequences in an analysis of covariance (ANCOVA) model with terms for baseline and treatment sequence.
Safety parametersAdverse events (serious and non-serious), concomitant drug and non-drug therapies, safety laboratory blood samples (hemoglobin, erythrocytes, hematocrit, thrombocytes, leukocytes, sodium, potassium, urea, creatinine, serum bilirubin level, aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, lactate dehydrogenase, follicle-stimulating hormone for postmenopausal women only), and urine samples (leukocytes, nitrite, urobilinogen, protein, pH, occult blood (erythrocytes, leucocytes), specific gravity, ketones, bilirubin, glucose) will be collected routinely throughout the study. Sparse pharmacokinetic blood sampling will be conducted in the Parent Study (visit 1–visit 6). Blood samples for the quantification of N-acetyl-L-leucine in plasma will be obtained at visit 7 and visit 9. Urine samples will also be collected for measuring concentrations of N-acetyl-D-leucine at the time points designated on the schedule of events (Suppl. Tables 1 and 2). At visit 1, this urine sample serves as a key enrollment criterion testing for the use of the prohibited medication N-acetyl-DL-leucine. Vital signs, physical exams, height/weight, and electrocardiograms will also be collected at the time points designated on the schedule of events (Suppl. Tables 1 and 2). A detailed description of the safety parameters is provided in Suppl. Material I.
All statistical analyses will be detailed in a separate statistical analysis plan. For each of the primary and secondary endpoints, there will be evaluations within key subgroups: naïve versus non-naïve as determined at screening; age (pediatric versus adult at the time of enrollment); age/weight/dosing group; age of diagnosis: early-infantile (< 2 years), late-infantile (2 to < 6 years), juvenile (6 to < 15 years), adolescent/adult (≥ 15 years); disease severity based on SARA below/above the median SARA score at visit 1; gender (male versus female); region (US versus rest of world); patients taking miglustat versus patients not taking miglustat; intra-patient variability between SARA score at visit 1 (baseline 1) versus visit 2 (baseline 2) (below/above median). These evaluations will be based on plotting treatment differences together with 90% confidence intervals within each subgroup.
In a one-sided test at the p = 0.05 level, a total sample size of 46 in a two-treatment, two-period placebo-controlled cross-over trial achieves approximately 80% power for treatment comparisons in relation to the SARA/mSARA total score assuming a true mean difference of 1.0/0.85 (respectively) and a standard deviation for the total SARA/mSARA score between 7.5 and 8.5 and 6.375 and 7.225 (respectively) based on an analysis of covariance with the baseline SARA/mSARA score at the start of period I as the covariate. These results, based on extensive simulations, assume a correlation of 0.95 between each of the pairwise outcomes: baseline for period I, endpoint for period I, and endpoint for period II. The values for the standard deviation and the correlations used in the calculation are guided by the results of the IB1001-201 study. These levels of power are maintained when there is a positive or negative period effect of up to 0.5 and 0.425 units on the SARA/mSARA scale, respectively.
A description of data collection is provided in Suppl. Material II.
留言 (0)