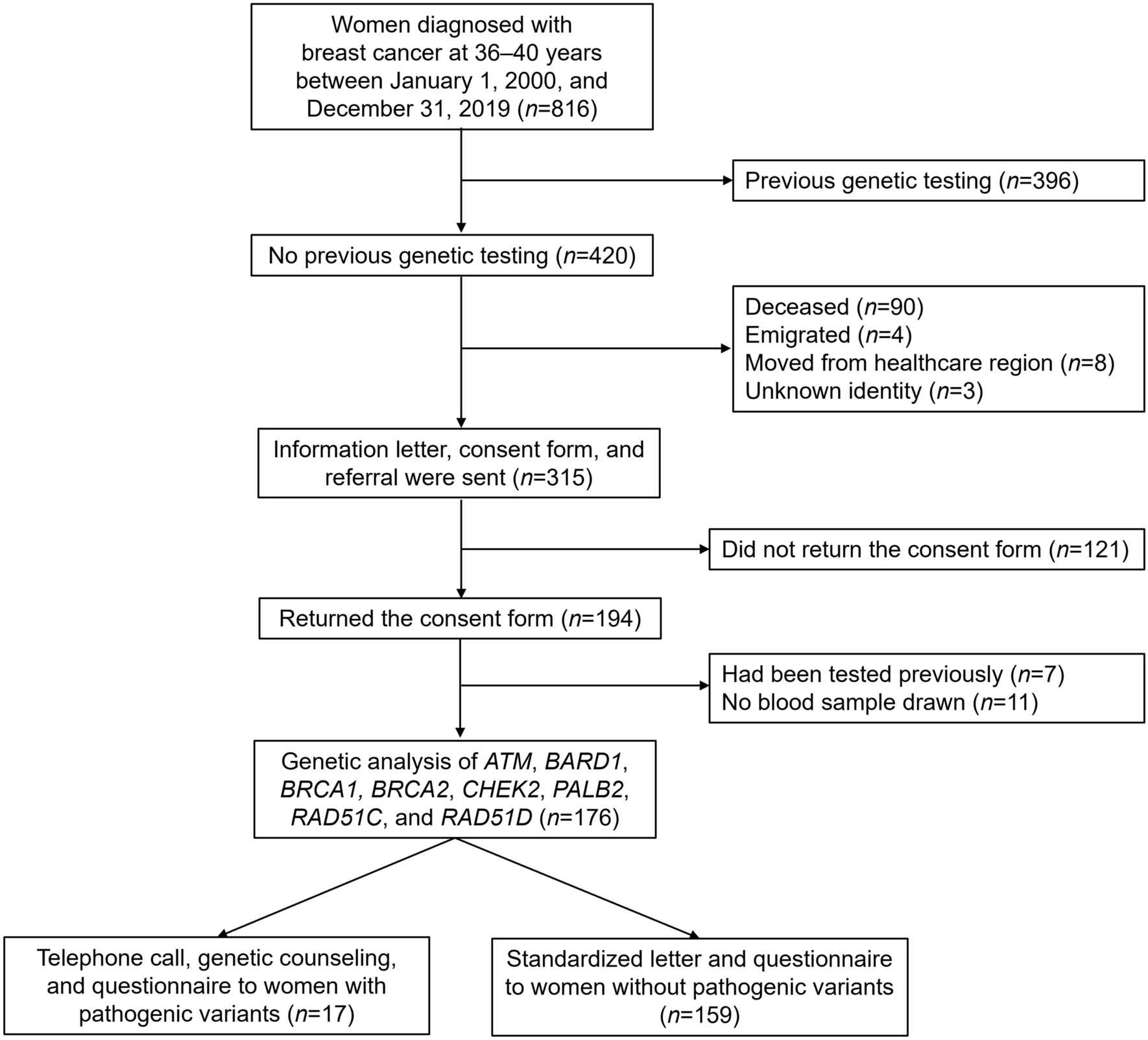
Cell cultures and reagents
Human TNBC cell lines: BT-549, HCC-1806, HCC-1937, MDA-MB-231, MDA-MB-453, MDA-MB-468 and SUM-159 were obtained from the American Type Culture Collection (ATCC) and were maintained using ATCC recommended media. All model cells utilized were free of mycoplasma contamination. Additionally, STR DNA profiling was used to confirm the identity of cell lines. The GAPDH, p-ERK (42/44), ERK (42/44), p-S6 (s235/236), S6, p-mTOR (S2448), mTOR, p-4EBP1, 4EBP1 and c-Myc antibodies were obtained from Cell Signaling Technology (Beverly, MA). LAS1L, TEX-10, SENP3 antibodies were purchased from Proteintech (Rosemont, IL). The β-Actin antibody (A-2066), WDR18 antibody (HPA050200) and Vinculin antibody (V9264) were purchased from Millipore Sigma (Burlington, MA). The Ki67 antibody (ab1667) was purchased from Abcam (Cambridge, MA). The PELP1 antibody (A300-180A) was purchased form Bethyl Laboratories Inc. (Montgomery, TX).
Cell viability, colony formation, and apoptosis assays
The effects of SMIP34 treatment on cell viability was assessed by using the MTT cell viability assay in 96-well plates as described [14]. Colony formation assays were done as described [14]. Briefly, BT-549 and MDA-MB-231 model cell lines (500 cells/well) were seeded in 6-well plates and allowed to grow for 5 days in control or SMIP34 treated medium and then medium was changed to normal medium and allowed to grow for 5–7 more days. Cells were then fixed in ice-cold methanol and stained with 0.5% crystal violet solution. The colony area percentage was calculated using NIH ImageJ software. Apoptosis was measured using Annexin V-PI staining (BioLegend, San Diego, CA) according to manufacturer’s protocol.
Cell invasion assays
The effect of SMIP34 on cell invasion of TNBC cells was determined by using the Corning® BioCoat™ Growth Factor Reduced Matrigel Invasion Chamber assay. MDA-MB-231 and BT-549 cells were treated with vehicle or SMIP34 (20 μM) for 22 h and invaded cells were determined according to manufacturer protocols.
Cell cycle and RT-qPCR analysis
TNBC cells were treated with either vehicle (0.1% DMSO) or SMIP34 (10 μM) for 48 h. Cells were then trypsinized and harvested in PBS, followed by fixation in ice-cold 70% ethanol for 30 min at 4 °C. Cells were washed again with PBS and stained with a mixture of propidium iodide (PI) and RNase A. The PI-stained cells were subjected to flow cytometry using a BD FACSCalibur™ (BD Biosciences). Total RNA extracted from the TNBC cells was used for real-time PCR. The real-time PCR primers that were utilized to validate the PELP1 target genes were purchased from Millipore Sigma (Burlington, MA).
Western blotting
TNBC model cells were subjected to cell lysis using either RIPA or NP-40/Triton X-100-lysis buffer containing protease and phosphatase inhibitors followed by Western blot analysis. For proteasome degradation experiments, MG132 was purchased from Sigma (Burlington, MA). After treatment, cells were subjected to cell lysis using NP-40/TritonX-100-lysis buffer containing protease/phosphatase inhibitors and deubiquitinating enzyme inhibitor N-Ethylmaleimide (NEM) (Selleck, Pittsburgh, PA) followed by Western blot analysis.
Immunohistochemistry (IHC)
IHC was performed as described previously [14]. Briefly, tumor sections were incubated with Ki67 (1:50) or PELP1 (1:150) primary antibody for overnight at 4 °C followed by secondary antibody incubation for 45 min at room temperature. Immunoreactivity was visualized by using the DAB substrate and counterstained with hematoxylin (Vector Lab, Burlingame, CA). A proliferative index was calculated as the percentage of Ki67 positive cells in five randomly selected microscopic fields at 20X per slide.
Ex vivo tumor studies
Excised tumor tissues from cell line-derived xenograft (CDX) and patient-derived xenograft (PDX) were processed, and cultured ex vivo as previously described [14]. Briefly, tissues were processed and excised into small pieces and cultured on gelatin sponges for 24 h in medium containing 10% FBS. Tissues were treated with vehicle or 20 μM SMIP34 in culture medium for 72 h and fixed in 10% buffered formalin at 4 °C overnight and subsequently processed into paraffin blocks. Sections were then processed for IHC of Ki67 staining.
In vivo orthotopic tumor models
All animal experiments were performed after obtaining VA and UTHSA IACUC approval. Female 8 weeks-old SCID or NSG mice were purchased from Jackson Laboratory (Bar Harbor, ME). For xenograft tumor assays, model cells (MDA-MB-231, 2 × 106) were mixed with an equal volume of Matrigel and injected into the mammary fat pads of female SCID mice as described [15]. For PDX studies, PDX tumor tissue was dissected into 2 mm3 pieces and implanted into the flanks of female NSG mice. When the tumor volume reached ~ 150 mm3, mice were randomized for treatment. Based on previous lab data as well as published findings, the numbers of animals needed were chosen to demonstrate differences in tumor incidence or treatment effect. Calculations are based on a model of unpaired data power = 0.8; p < 0.05. Once tumors reached measurable size (~ 150–200mm3), mice were divided into control and treatment groups (n = 7 or 8 tumors per group). The control group received vehicle and the treatment groups received SMIP34 (20 mg/kg/i.p./5 days/week) in 0.3% hydroxypropyl cellulose. Animals were monitored daily for adverse toxic effects. The TM89 and TM96 PDX models were purchased from Jackson Laboratory. Tumor growth was measured by caliper at 3–4 days intervals. At the end of each experiment, animals were euthanized, and the tumors excised, weighed, and processed for IHC staining.
Statistical analyses
Statistical differences between groups were analyzed with unpaired Student’s t-test and ANOVA using GraphPad Prism 9 software. All the data represented in plots are shown as means ± SE. A value of p < 0.05 was considered as statistically significant.






留言 (0)