
記住我






 Figure
Figure Box 1
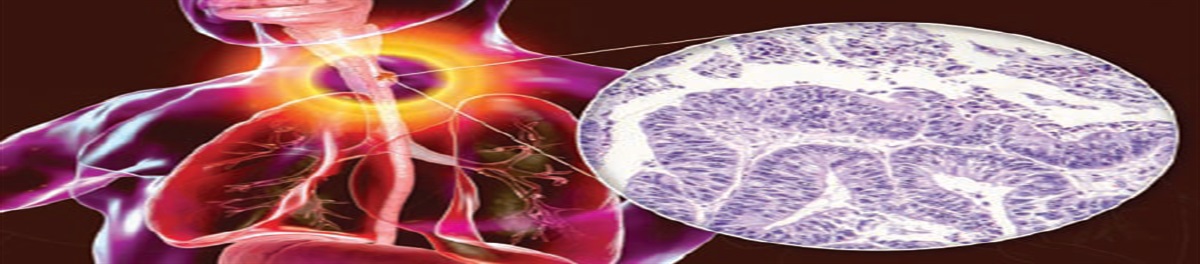
Box 1Myelodysplastic syndromes (MDS) are disorders associated with dysfunction of blood cell maturation, dysplastic cells, and cytopenia.1 More than 45,000 new cases of MDS are diagnosed annually, and the risk increases with age, male sex, and White ethnicity.2,3 The majority of patients with MDS are over age 69 years.3 MDS results in the formation of immature blood cells and affects one or more cell lines, resulting in anemia, neutropenia, and/or thrombocytopenia (Figure 1). Initial suspicion for MDS may be prompted if a patient has symptoms of anemia such as fatigue or dizziness, or cytopenia on routine complete blood cell (CBC) count. Unexplained cytopenia, multiple cytopenias, and persistent anemia without a cause should increase suspicion and prompt a referral to hematology.1 Bone marrow examination, including molecular and cytogenetic analysis, is needed to establish the diagnosis. Treatment focuses on managing cytopenia and identifying patients eligible for allogeneic stem cell transplant, the only curative treatment option.4 Primary care providers (PCPs) should be prepared to identify and manage common complications associated with cytopenia, such as infection and bleeding, and be aware that about 30% of patients progress to acute myelogenous leukemia (AML).1
 FIGURE 1.: MDSReprinted with permission from the Centre for Clinical Haematology, Singapore. https://cfch.com.sg.
FIGURE 1.: MDSReprinted with permission from the Centre for Clinical Haematology, Singapore. https://cfch.com.sg.Identification, treatment, and prognostic indicators for MDS have improved significantly over the past 20 years, especially with the implementation of the 2016 World Health Organization (WHO) classification system and the 2012 Revised International Prognostic Scoring System (IPSS-R).5,6 These guidelines have led to early identification, diagnosis, and implementation of therapeutic measures that may improve patient outcomes and quality of life. Therefore, PCPs treating patients with suspected MDS should focus on early hematology referral. This article summarizes the pathophysiology, clinical presentation, and treatment of MDS from a primary care perspective.
PATHOPHYSIOLOGYMDS arises from clonal mutation of stem cells that are defective, immature, and have a competitive advantage over normal stem cells. Mutations occur secondary to genetic susceptibility or damage to hematopoietic cells.1 This leads to the replacement of normal bone marrow with mutated cells, resulting in cytopenia of one or more cell lines.7
In about 80% of patients, MDS is idiopathic.1 Secondary causes include exposure to radiation, chemotherapy, heavy metals, toxic chemicals, viral infections, and autoimmune diseases. PCPs should be aware of the increased risk of developing therapy-related MDS in patients with a history of radiation and/or chemotherapy. Therapy-related MDS often is associated with the use of alkylating agents (cisplatin and carboplatin, used to treat testicular and ovarian cancer) and purine analogues (fludarabine and azathioprine, used to treat leukemia and autoimmune disease).8 Therapy-related MDS has a poor prognosis, and patients should be closely monitored for progression to AML.9
 Box 2
Box 2About 20% of patients diagnosed with MDS have autoimmune disorders, and MDS should be considered in patients with rheumatologic, inflammatory, or immune-related symptoms, especially if they also have cytopenia.8 A growing number of autoimmune disorders—ranging from limited disease to multiorgan systemic illness—are associated with MDS (Table 1).10-12 Autoimmune disorders commonly associated with MDS include vasculitis, connective tissue disease, and inflammatory arthritis.13
TABLE 1. - Autoimmune disorders linked to MDS10-12Connective tissue disorders
Vasculitis
Neutrophilic dermatosis
Sweet syndrome
Pyoderma gangrenosum
Inflammatory arthritis
Other
The signs and symptoms of MDS are related to cytopenia. Some patients are asymptomatic, with abnormalities identified only on routine CBC count.8 Macrocytic anemia is identified in 90% of patients and is a key finding.8 Patients with anemia may present with symptoms such as fatigue, weakness, angina, or dizziness. Corresponding physical examination findings include pallor, tachycardia, or hypotension. Thrombocytopenia occurs to a lesser extent than anemia and is present in 40% to 65% of patients with MDS.14 Decreased platelet production may result in easy bruising or bleeding and physical examination findings such as petechiae, ecchymosis, purpura, or bleeding gums. Neutropenia is also less common than anemia but is found in 30% of patients with MDS, who may present with persistent infection or fever.8 Although the risk varies with severity and duration, neutropenia increases patients' risk for infection–-especially infection caused by Gram-negative bacilli, Gram-positive cocci, or fungi.8,15 Consult the Infectious Diseases Society of America's guidelines for evaluating and managing patients with neutropenia.15
DIAGNOSTIC TESTINGDiagnosis and classification of MDS is based on evaluation of a CBC count with blood smear and bone marrow examination.1 In addition to assessing the patient's clinical presentation, diagnosis and classification center on the number and type of cytopenia(s), cell dysplasia on morphologic examination, and identification of chromosomal abnormalities on cytogenetic and molecular genetic analysis of the peripheral blood and bone marrow.16 Other causes of cytopenia or dysplasia, such as toxins, autoimmune diseases, nutritional deficiencies, and various infections, must be ruled out to make the diagnosis of MDS. Initial diagnostic studies include serum erythropoietin, iron, ferritin, total iron-binding capacity, vitamin B12, thyroid-stimulating hormone, lactate dehydrogenase, and HIV testing.1
Under proposed diagnostic criteria for MDS, a patient must have persistent cytopenia for more than 4 months in one or more of the cell lines.16 Macrocytic anemia often can be identified by evaluating the peripheral blood via a CBC count with differential and blood smear. Thrombocytopenia and/or leukopenia increase suspicion for MDS.1 Isolated thrombocytopenia or leukopenia are uncommon.
Hypercellularity and morphologic cellular dysplasia identified by bone marrow examination (Figure 2) are key findings in MDS and may include abnormalities in singular or multiple lines (erythroid, myeloid, and megakaryocyte).5 Morphologic dysplasia of 10% or more in one or more of the cell lines is the diagnostic threshold.5 The percentage of blast cells differentiates MDS (less than 20%) from AML (greater than 20%).16
 FIGURE 2.:
FIGURE 2.: Bone marrow aspirate smear with dysplastic erythroid cells
Cytogenetic and molecular genetic analysis, including fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), and next-generation sequencing, may be used to identify chromosomal abnormalities or gene mutations. Cytogenetic abnormalities have been reported in 30% to 50% of patients with MDS.17 Although a wide range of genetic abnormalities are associated with MDS, the most common mutations are +8, +9, -7, del(7q), del(20q), del(13q), and isochromosome 17q.17 As defined by the WHO classification, several chromosomal abnormalities can be considered presumptive of MDS in patients with persistent cytopenia, even in the absence of diagnostic morphologic changes.5,18
PROGNOSTIC INDICATORSClassification of MDS has been revised to combine the 2016 WHO criteria, National Comprehensive Cancer Network (NCCN) criteria, and the 2012 IPSS-R; the result is five risk categories: very low, low, intermediate, high, and very high.6 This classification helps clinicians initiate treatment and determine how aggressively to treat patients. Classification is based on patients' cytogenetic evaluation, percentage of bone marrow blasts, degree of anemia, presence of thrombocytopenia, and degree of neutropenia; patients with higher scores have worse outcomes. The degree of neutropenia, degree of thrombocytopenia, and percentage of blasts in the bone marrow can be used to estimate the risk of AML conversion and prognosis.9 Patients classified in the very low-risk group have an overall median survival of about 8.8 years combined with a low probability of conversion to AML (average 14.5 years).6 In contrast, patients in the very high-risk group have a median survival of less than 1 year and a high probability of conversion to AML (average 0.7 years).6
TREATMENT APPROACHTreatment of MDS incorporates the 2016 WHO criteria, NCCN criteria, and 2012 IPSS-R prognostic scoring, along with clinical guidance from the hematologic and transplant teams.19Stem cell transplant offers a potential cure, and the remaining management options focus on correcting specific cytopenias, reducing symptoms, and improving patient comfort. Active surveillance is appropriate for patients who are asymptomatic and at low risk, and for higher-risk patients with poor prognostic indicators.19 Correction of cytopenias using erythropoiesis-stimulating agents (ESAs), hypomethylating agents, immunomodulatory agents, myeloid growth factors, transfusion, and hematopoietic stem cell transplantation are summarized in the following sections.
Anemia treatment optionsMore severe anemia and higher frequency of RBC transfusion are included in the 2016 WHO classification as poor prognostic indicators.1 Patients with symptomatic anemia can be treated with ESAs such as epoetin to stimulate RBC production; with an adequate response, maintenance doses can be administered with varying frequency and dosing.1,7 In general, ESA use may improve cell line production by 39% and is particularly beneficial in low-risk patients with symptomatic anemia.1 Higher-dose ESAs can boost erythroid production by 60%.4 Some patients with MDS may not respond to ESAs and remain dependent on transfusions.7 Patients with transfusion-dependent anemia must be closely monitored by a hematologist and assessed for iron overload.
Hypomethylating agents such as azacitidine and decitabine display antitumor effects and may enhance patient quality of life by improving cytopenia by 25% to 40% and reducing transfusion-dependent anemia by 16%.8 These agents are beneficial in high-risk patients who do not qualify for transplant or those who have relapsed despite transplant.20 Hypomethylating agents cause a variety of adverse reactions, ranging from gastrointestinal disturbance to significant myelosuppression.19,21
Immunomodulatory agents such as lenalidomide have been found to reduce transfusion dependence in 26% of patients without chromosome 5q deletion MDS (non-del[5q]MDS) and may be instituted in those with poor response rates to ESAs.4 Luspatercept is approved for use in some low-risk patients with refractory MDS. This drug has been shown to improve quality of life and reduce transfusion dependence by 10% in patients with non-del(5q) MDS and 33% in those with del(5q) MDS, and has a similar adverse reaction profile to hypomethylating agents.20
PCPs should maintain realistic expectations for patient hemoglobin levels—10 to 12 g/dL, and not exceeding 12 g/dL.4 Collectively, the care team should be aware of thromboembolic events that are associated with the use of ESAs. PCPs can assist specialists by managing coexisting causes of anemia such as nutritional deficiencies, renal insufficiency, or gastrointestinal blood loss.
Neutropenia treatment optionsNeutropenia is present in 20% to 25% of patients with MDS; patients with an absolute neutrophil count of less than 1,000 cells/mm3 are considered at risk for infection.21 Myeloid growth factors such as granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor often are used to boost neutrophil production.8 According to NCCN, these agents are not recommended for routine infection prophylaxis and administration should be reserved for patients with a history of recurrent or resistant infections.4 Prophylactic antibiotics such as levofloxacin and antifungal agents such as posaconazole cover pathogens commonly seen in patients with neutropenia such as Gram-negative bacilli, Gram-positive cocci, and fungi (Candida and Aspergillus). Antibiotic prophylaxis should be used in patients at risk of infection who do not qualify for myeloid growth factors.7 Myeloid growth factors produce mild to moderate bone pain in 10% to 30% of patients and discomfort at the injection site.21
Thrombocytopenia treatment optionsThrombocytopenia has been defined by the 2012 IPSS-R as a platelet count below 100,000 cells/mm3.1 Platelet transfusions are the standard management of correcting thrombocytopenia in patients with MDS; however, most patients do not reach critically low levels requiring transfusion (counts less than 10,000 cells/mm3 or active signs of ominous bleeding with a platelet count below 100,000 cells/mm3).1 Thrombopoietin receptor agonist medications such as romiplostim and eltrombopag are being explored as treatment options but should be used with caution because of the risk of blast proliferation and acceleration to leukemic transformation.1 Patients who are dependent on transfusions or whose disease does not respond to treatment should be screened for full human leukocyte antigen typing, as should the blood donor.1 Use irradiated blood products to reduce the incidence of transfusion-related graft-versus-host disease.22
Stem cell transplantationAllogeneic hematopoietic stem cell transplantation is the process of transferring stem cells to a patient from a healthy donor and is considered the only potential cure for MDS.4 The NCCN reported 5-year transplant survival rates of 40% in high-risk patients and 65% in intermediate-risk patients.5 Unfavorable outcomes occur with increased age, disease burden, and percentage of marrow blasts. Patients age 60 years or younger and in higher-risk categories have increased life expectancy when transplant occurs shortly after diagnosis.19 Emerging evidence discourages the use of advanced age as the sole excluding factor for transplant.20 However, clinicians must be aware of the higher morbidity and mortality in older adults.7 Treatment decisions are dictated by the transplant team, hematologist, and institutional policies.
CONCLUSIONDiagnosis and management of MDS is complex and requires an integrated approach, including prompt referral to a hematologist for further evaluation. Patients with unexplained cytopenia and unexplained or refractory anemia should be worked up for MDS and assessed for risk of AML conversion. Diagnosis and treatment are driven by results of peripheral blood, bone marrow, and cytogenetic analysis. Combining formal classification systems, prognostic criteria, and evaluation of the patient holistically assists in personalizing treatment therapy and improving outcomes. Treatment requires specialty care, but PCPs must monitor for complications such as medication-related thromboembolic events, acute bleeding episodes, and screening for neutropenia-related infections.
REFERENCES 1. Mohammad AA. Myelodysplastic syndrome from theoretical review to clinical application view. Oncol Rev. 2018;12(2):397. 2. Menssen AJ, Walter MJ. Genetics of progression from MDS to secondary leukemia. Blood. 2020;136(1):50–60. 3. National Cancer Institute. Surveillance, epidemiology, and end results program. https://seer.cancer.gov/statistics-network/explorer/application.html. Accessed March 18, 2023. 4. Montoro J, Yerlikaya A, Ali A, Raza A. Improving treatment for myelodysplastic syndromes patients. Curr Treat Options Oncol. 2018;19(12):66. 5. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. 6. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. 7. Cazzola M. Myelodysplastic syndromes. N Engl J Med. 2020;383(14):1358–1374. 8. Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014;383(9936):2239–2252. 9. Kuendgen A, Nomdedeu M, Tuechler H, et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification-an approach to classification of patients with t-MDS. Leukemia. 2021;35(3):835–849. 10. Grignano E, Jachiet V, Fenaux P, et al. Autoimmune manifestations associated with myelodysplastic syndromes. Ann Hematol. 2018;97(11):2015–2023. 11. Albert DA, Burns CM. Rheumatologic manifestations in myelodysplastic syndrome. J Clin Rheumatol. 2012;18(3):148–150. 12. Fozza C, Crobu V, Isoni MA, Dore F. The immune landscape of myelodysplastic syndromes. Crit Rev Oncol Hematol. 2016;107:90–99. 13. Wolach O, Stone R. Autoimmunity and inflammation in myelodysplastic syndromes. Acta Haematol. 2016;136(2):108–117. 14. Tsagarakis NJ, Paterakis G, Papadhimitriou SI, et al. Bone marrow aspirate automated counts on hematology analyzers: formulating a scoring system based on hematology parameters, to discriminate reactive versus myelodysplastic syndrome-related bone marrows. Int J Lab Hematol. 2019;41(4):542–549. 15. Freifeld AG, Bow EJ, Sepkowitz KA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2011;52(4):e56–e93. 16. Valent P, Orazi A, Steensma DP, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8(43):73483–73500. 17. Cross NCP, Godfrey AL, Cargo C, et al. The use of genetic tests to diagnose and manage patients with myeloproliferative and myeloproliferative/myelodysplastic neoplasms, and related disorders. Br J Haematol. 2021;195(3):338–351. 18. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. 19. Platzbecker U. Treatment of MDS. Blood. 2019;133(10):1096–1107. 20. Bewersdorf JP, Carraway H, Prebet T. Emerging treatment options for patients with high-risk myelodysplastic syndrome. Ther Adv Hematol. 2020;11:1–22. 21. Shallis RM, Zeidan AM. Management of the older patient with myelodysplastic syndrome. Drugs Aging. 2021;38(9):751–767. 22. Loingsigh SN, Flegel WA, Hendrickson JE, Tormey CA. Preventing transfusion-associated graft-versus-host disease with blood component irradiation: indispensable guidance for a deadly disorder. Br J Haematol. 2020;191(5):653–657.
留言 (0)