Cell lines and cell culture
The human colon cancer cell lines HCT116 (wild-type p53) and HT29 (mutant p53; R273H) were purchased from ATCC. Cells were maintained in RPMI-1640 (PAN Biotech P04-18,500) supplemented with 10% fetal bovine serum (FBS; PAN Biotech P30-3306) and 1% penicillin‒streptomycin (P/S; PAN Biotech P06-07,100). All cells were cultured at 37 °C with 5% CO2. CRISPR-Cas9 genome editing was used to stably knock out the ATF2 gene in HCT116 (designated clones named E5 and F9) and HT29 (designated clones named B5 and F10) cells. The validation of the ATF2 knockout and functional description of the newly generated ATF2 knockout cell lines are given in Huebner et al. (2022) [4]. HCT116 p53−/− cells were generously provided by Bert Vogelstein (Johns Hopkins) and maintained in McCoy’s 5A medium (Life Technologies, Darmstadt, Germany) containing 10% FBS and 1% P/S. The three cell lines were authenticated using Multiplex Cell Authentication by Multiplexion (Heidelberg, Germany). Mycoplasma-free status was verified.
Drugs and chemicals
The autophagy inhibitor bafilomycin A1 (B1793-10 µg) was purchased from Sigma‒Aldrich and dissolved in 100% dimethyl sulfoxide (DMSO, Pan Biotech P6036720100) to a 16 μM stock solution; the Chk1 inhibitor (PF-477736, PZ0186) was purchased from Sigma‒Aldrich and dissolved in DMSO to a 10 mM stock solution; the JNK inhibitor (SP600125, tlrl-sp60) was purchased from InvivoGen and dissolved in DMSO to a 50 mM stock solution; and Z-VAD-FMK (S7023) was purchased from Selleckchem and dissolved in DMSO to a 10 mM stock solution. The single compound 5-fluorouracil (5-FU) was purchased from Sigma‒Aldrich and dissolved in DMSO to a 100 mM stock solution. All stocks were stored at − 20 °C.
Western blotting
Western blotting analysis was performed as described previously [21]. Briefly, 20–60 μg protein was loaded into SDS‒PAGE gels and transferred onto 0.2 μm nitrocellulose membranes (GE Healthcare, 10,600,006) overnight. After blocking with 5% milk (Carl Roth, T145.2) in TBST for 1 h at room temperature, the membranes were incubated with corresponding primary antibodies overnight at 4 °C: caspase 9 (Cell Signaling, 9502, 1:1,000), Bcl-2 (Cell Signaling, 2872, 1:1,000), Bax (Cell Signaling, 5023, 1:1000), H2AX (Millipore, 07–627, 1:10,000 – 1:15,000), γ-H2AX (phospho-Ser139, Millipore, 05–636, 1:5,000 – 1:7,000), p-ATF2 E268 (p-Thr71, Abcam, ab32019, 1:5000), ATF2 E243 (Abcam, ab32160, 1:10,000), p-SAPK/JNK (p-Thr183/Tyr185, Cell Signaling, 4668, 1:1,000), SAPK/JNK (Cell Signaling, 9258, 1:1,000), p-p38 (p-Thr180/Tyr182, Cell Signaling, 9211, 1:1,000), p38 (Cell Signaling, 9212, 1:1,000), p-p44/42 (ERK1/2)(p-Thr202/Tyr204, Cell Signaling, 9101, 1:1,000), p44/42 (ERK1/2, Cell Signaling, 9102, 1:5,000), PARP (Cell Signaling, 9532, 1:1,000), p62 (Cell Signaling, 5114, 1:1,000), p-Chk1 (p-Ser317, Cell Signaling, 2344, 1:1,000), Chk1 (Santa Cruz, sc-8408, 1:500), p-ATR (p-Thr1989, Cell Signaling, 30,632, 1:1,000), ATR (Cell Signaling, 13,934, 1:1,000), and secondary antibodies (anti-mouse and anti-rabbit IgG peroxidase conjugated, Pierce, Rockford, IL, USA, 1:10,000), GAPDH-HRP (Abnova, MAB5476, 1:50,000–1:100,000). Experiments were performed in independent biological duplicates. For Western blot quantification of -H2AX/H2AX, p-Chk1Ser317/Chk1, and p-ATRThr1989/ATR, band intensities were quantified by ImageJ (National Institute of Health, USA) and normalized to each corresponding GAPDH (housekeeper). Ratios were calculated by dividing phosphorylated protein of interest by nonphosphorylated protein of interest. Ratios for cleaved PARP were determined by ImageJ, but ratios were calculated by dividing cleaved PARP by noncleaved PARP.
Co-Immunoprecipitation (Co-IP)
Co-IP was performed according to the ‘Dynabeads® Protein G Immunoprecipitation Kit’ manual (Thermo Fisher, 10007D) with slight adjustments. Briefly, protein lysate (500 μg/sample) was adjusted to a final volume of 200 μl and incubated with primary antibodies (ATF2 E243 (Abcam, ab32160, 1:50), p53 (Santa Cruz, sc-126, 1:20), Chk1 (Santa Cruz, sc-8408, 1:20)) overnight on rotation at 4 °C. The next day, the mixture was incubated with 30 µl Dynabeads® for 15 min at RT, which had been prewashed once with 200 µl Ab-Binding & Washing Buffer. Immunocomplex was then washed with 200 µl Washing Buffer 3 times. The Dynabeads®-Ab-antigen complex was resuspended in 100 µl Washing Buffer and transferred to new Eppendorf tubes. The supernatant was removed, 20 µl elution buffer and 4 µl SDS (6X) loading buffer were added, and the samples were subjected to SDS‒PAGE. Western blotting was performed following standard procedures.
Chorioallantoic membrane (CAM) assay and immunohistochemistry
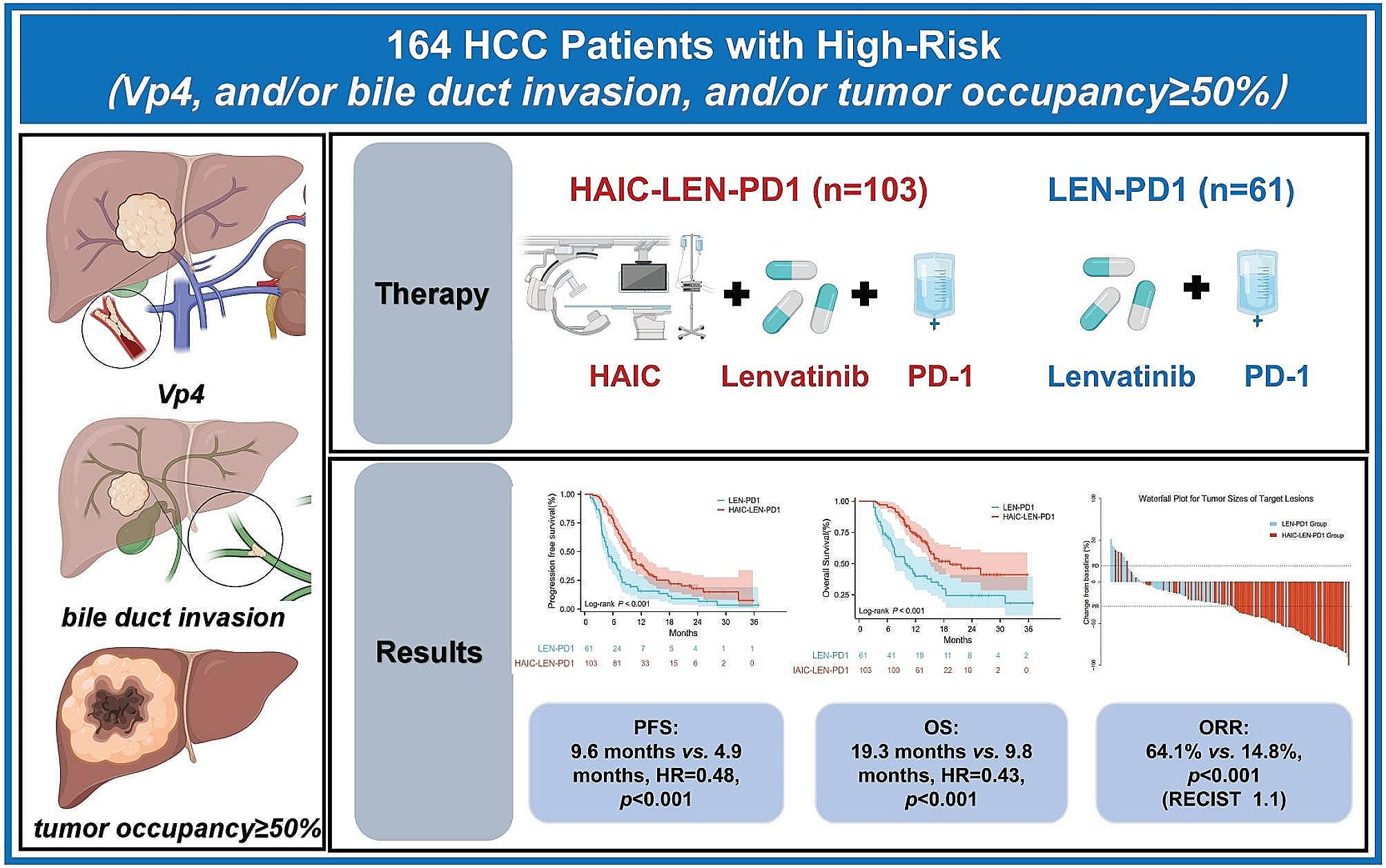
Fertilized specific-pathogen-free eggs (Valo Biomedia) were incubated at a temperature of 37 °C and constant air humidity of 70%. Eggs were opened on day 8 of chicken embryo development, and the hole was sealed with surgical tape. On day 9, 1.0 × 106 cells pretreated with 15 μM 5-FU for 48 h were resuspended in a mixture of 20 µl RPMI 1640 medium and 20 µl Matrigel. The 40 µl cell pellet was slowly placed onto the CAM. After incubating for 5 days, xenografts with surrounding CAM were harvested and fixed in 4% paraformaldehyde for 24 h before paraffin embedding. Serial slides were stained with H&E and antibodies against ATF2 (E243, Abcam 1:100,000), Ki67 (Dako, 1:100), pan cytokeratin (Zytomed, 1:40), p-Chk1Ser317 (Abcam, 1:2,000), and γ-H2AX (Abcam, 1:2,000).
RNA Interference
Interfering ON-TARGETplus SMARTpool ATF2 siRNA (D-009871–00-0005) and scramble control siRNA (ON-TARGETplus Nontargeting pool, D-001810–10-05) were purchased from Dharmacon. Transfection was performed in 6-well plates using Lipofectamine RNAiMAX reagent (Thermo Fisher, 13,778,075) in OptiMEM (Thermo Fisher, 31,985,062) containing 25 pmol of siRNA/scramble in each well. Cells were counted and seeded again after 48 h of transfection at 37 °C and 5% CO, followed by 48 h of treatment with 5-FU. Knockdown efficiency was assessed by Western blotting.
Colony formation assay
Cells were pretreated with 15 µM 5-FU or DMSO. After 48 h of incubation, the cells were trypsinized, and 1000 cells were seeded in a 6 cm plate with normal growth medium and incubated for an additional 10 days (control group) and 20 days (treatment group). As the colonies became visible, the cells were fixed and permeabilized with 70% methanol for 20 min. Afterwards, colonies were stained with crystal violet for 20 min, washed with water and allowed to dry. Pictures were taken with a digital camera, and the number of colonies was recognized by Image-Pro Plus software.
Annexin-propidium iodide apoptosis assay
Detection of apoptosis was performed by Annexin-PI staining as described previously [22]. Cells were treated with 15 μM 5-FU for 48 h or with normal medium for 24 h. The fluorescent signal was measured by flow cytometry (BD FACSCanto® II, BD Biosciences). The data were evaluated using FlowJo 7.6.5 software.
Immunofluorescence
Cells were seeded on coverslips and treated with 15 μM 5-FU or DMSO for 48 h. After the treatment, the cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 in PBS for 10 min. Afterwards, the cells were blocked with 3% BSA in PBS for 30 min on a shaker and subsequently incubated with primary antibody (p-ATF2 E268 (p-Thr71, Abcam, ab32019, 1:500)), which was diluted in 3% BSA in PBS for 1 h at room temperature, and secondary antibody (goat anti-rabbit IgG, Alexa Fluor 555, Thermo Fisher, A-21428, 1:500), which was diluted in 1% BSA for 1 h in a dark room. Nuclei were stained with DAPI (Sigma‒Aldrich, MBD0015-5ML, 1:1,000) in PBS for 25 min. Fluorescence images were taken by a Nikon Ti-S fluorescence microscope.
Duolink® Proximity Ligation Assay (PLA)
Cells were seeded onto an Ibidi µ-Slide chamber. After 48 h of treatment with 5-FU or DMSO, cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton-X for 15 min. Thereafter, the cells were blocked with 3% BSA for 30 min and incubated with a mixture of primary antibodies for 1 h (p-ATR (p-Thr1989, Thermo Fisher, MA5-27,731, 1:500) and ATF2 E243 (Abcam, ab32160, 1:500)) diluted in 3% BSA. Then, the cells were incubated with secondary antibody for 1 h in a humidity chamber at 37℃ (mixture of Duolink® In Situ PLA® Probe Anti-Mouse MINUS (Sigma‒Aldrich, DUO92001-30RXN) and Duolink® In Situ PLA® Probe Anti-Rabbit MINUS (Sigma‒Aldrich, DUO92005-30RXN)). Hybridization, ligation, amplification, and detection were performed according to the manufacturer’s instructions (Duolink™ In Situ Detection Reagents Red, Sigma‒Aldrich, DUO92008). Finally, cells were stained with DAPI (1:1,000) in 0.01 × Wash Buffer B for 10 min before acquiring images. The technical negative control was incubated with PLA secondary antibody; the positive control was incubated with a mixture of p21/p-Chk2Thr68 primary antibodies (p21 (Cell Signaling, 2946, 1:400) and p-Chk2 (p-Thr68, Cell Signaling, 2197, 1:200)). PLA foci were evaluated by the software BlobFinder [23]. Nuclei and PLA foci were automatically detected by the software to allow quantification of PLA foci (nucleus and cytoplasm) per cell. Signals detected outside of cells were categorized as background and served for background correction.
Modelling of the ATR-Chk1 complex
Energy-minimized models of ATR, ATF2 and Chk1 were used to develop the complexes. The ATR-ATF2 complex was generated by docking individual protein structures using the protein‒protein docking server ClusPro [24] (https://cluspro.org/home.php). The cluster scores of the complexes from ClusPro were utilized to select the most reliable ATR-ATF2 complex. Furthermore, the interactions (hydrogen bonds) between the proteins were calculated using the Protein Interaction Calculator [25]. Similarly, the ATR-Chk1 complex was also developed using the ClusPro server, and the interaction was calculated using the Protein Interaction Calculator.
Modelling of the (ATR-ATF2)-Chk1 complex
This was further used to dock the ATR-ATF2 complex with Chk1 to develop the ATR-ATF2-Chk1 complex. Energy of the developed models and complexes computed using the Gromos96 force field [26]. Further active site pockets of ATR and ATF2 were predicted using the CASTp online server [27]. The interacting active site residue region of ATR and ATF2 was knocked out from the complex using Pymol [28], further protein‒protein docking was carried out using the ClusPro server, and interactions were calculated.


留言 (0)