
記住我


Macroscopic autopsy findings reported during tissue dissection, histological examination findings, and phenotypic associations are provided for the most prevalent and some of the unusual cases and disease entities seen (Table 3).
Table 3 Main autopsy findings and phenotypic associations of the most prevalent entitiesInflammationThe nature of the inflammation in the majority of the myocarditis cases was lymphocytic, secondary to an acute viral infection. Enteroviridae were the most prevalent viruses demonstrated in myocarditis (15 cases) with Coxsackie-B sub-type (including types 3, 4) and Coxsackie-A (type 10) isolated from ventricular tissue following RT-PCR sequencing.
There was one case of an 18 months toddler with infective aortitis and endocarditis where human herpes virus-6 DNA was identified in association with Coxsackie-A (type 10) RNA from myocardial tissue. Macroscopic examination of the heart revealed a bacterial endocarditis obliterating the non-coronary leaflet with the formation of an abscess around the root of the aorta. The heart had a normal weight (52 gm). There was evidence of atrial inflammation which extended into the sinoatrial node. Staphylococcus aureus, cultured from the root of the aorta, resulted in aortic valvular destruction with vegetations, which on histology showed fibrin and neutrophilic infiltrate. Other viruses isolated included Parvovirus-B19 (3 cases). An unusual case corresponded to a 13 years old female, who has been admitted with dyspnea, abdominal pain, and pre-syncopal episodes. At post-mortem, the heart was globular and enlarged weighing 302 g (normal heart weight: 154 gm) with evidence of left ventricular subendocardial fibroelastosis, cardiac myocyte hypertrophy, and patchy ventricular fibrosis on histology. Symptoms in life were diagnosed as probable viral gastroenteritis 11 days prior to death. Although the histology did not identify a prominent lymphocytic myocarditis, Parvovirus-B19 infection was investigated with PCR, confirming its high viral load in myocardial tissue [7].
Parainfluenza type 3 infection was identified in myocardial tissue using PCR in 3 cases of dilated cardiomyopathy including one case in an 8-month-old infant with 2 days of central cyanotic episodes preceding death. This association has been rarely reported in pediatrics in the published literature [8].
In 4 cases of myocarditis, no infectious isolate was identified despite a lymphocytic histological picture post-mortem.
Bacterial isolates in myocarditis included one case of group A streptococcus and one case of mycoplasma pneumoniae myocarditis in a girl admitted with lower respiratory tract infection who deteriorated rapidly and was found to have a co-morbid atrioventricular septal defect at post-mortem.
Six cases of encephalomyocarditis were identified in the study, all of which were due to Enteroviridae (Coxsackievirus-B2), sequenced from cerebrospinal fluid and myocardial tissue. Macroscopically, the cases showed cerebral congestion, edema, and cerebral necrosis, as well as an enlarged heart with mottled endocardium (Fig. 2a). Histological findings included foci of necrosis associated with lymphoid inflammatory infiltrate in the myocardium, leptomeninges, and cerebrum, and particularly prominent in the brain stem. This latter is characteristically most severely affected in enterovirus encephalitis, predominantly the ventral, medial, and caudal areas [9] (Fig. 2b–e). All cases presented with seizures.
Fig. 2
A few days old newborn who died suddenly. The post-mortem identified encephalomyocarditis caused by Coxsackie virus Type 3: a left ventricle with mottled appearances of the myocardium with pale and hemorrhagic areas; b myocardial necrosis associated to lymphocytic infiltrates and occasional eosinophils (H&E × 200); c C9 immunostain confirms extensive myocardial necrosis (× 100); d widespread encephalitis (H&E medulla × 200); e myelitis (H&E cervical spinal cord × 200)
Eosinophilic myocarditis was present in 4 cases with no previous medical history of note. The heart was morphologically normal; histology showed an eosinophilic-rich inflammatory infiltrate in the myocardium with myocyte necrosis and interstitial fibrosis. A specific preceding factor resulting in the hypersensitivity reaction was not found (Fig. 3).
Fig. 3
Eosinophilic (hypersensitivity) myocarditis in a teenager male found dead in the morning. The heart showed myocardial necrosis, patchy areas of interstitial fibrosis, intercellular edema, and numerous eosinophilic infiltrates (left ventricle H&E × 200). No organisms were recovered from the myocardial tissue culture
CardiomyopathiesIn total, 32 cardiomyopathies presented as a sudden and unexpected death. Of these, 12 were hypertrophic, and 5 presented as dilated cardiomyopathy. Interestingly, molecular investigations in one of the cases with dilated cardiomyopathy unveiled a previous Parvovirus B19 infection as the primary etiology. There were a few examples of very rare cardiomyopathies (Table 1): histiocytoid, non-compaction, mitogenic, cardiomyopathy in association with GM1 gangliosidosis, Beckwith-Wiedemann Syndrome, and myotonic dystrophy. Four cases presented with subendocardial fibroelastosis as the main finding at post-mortem, and when electron microscopy was conducted, these cases were considered to be related to mitochondrial abnormalities. The post-mortem investigation did not include specific genetic analysis nor next-generation sequencing. In some cases, the presence of an associated condition allowed a diagnosis (i.e., GM1 gangliosidosis, Beckwith-Wiedemann Syndrome, or myotonic dystrophy). In other cases, the presence of specific histological features favored a specific cardiomyopathy (i.e., histiocytoid, mitogenic, or non-compaction cardiomyopathy). Some of the rarest cases are summarized below:
A 1-month male infant born at normal gestational age was found apneic lying in a prone position. PM findings showed left ventricular non-compaction, a mitochondrial X-linked heritable condition. The heart weight was normal (23.7 gm). Histological abnormalities included enlarged myocyte nuclei with expanded cytoplasm. This condition has been reported to have phenotypic associations with mosaic trisomy 22 [10].
A 19-month male infant born to consanguineous parents with a history of intrauterine growth restriction and a sudden movement deterioration was admitted to the hospital with bradycardia. Echocardiography revealed a dilated left atrium and ventricle, tricuspid regurgitation, and mitral regurgitation. The patient subsequently suffered a pulseless electric activity (PEA) arrest. Macroscopic and microscopic examination confirmed subendocardial fibroelastosis in the left ventricular endocardium. The heart weight (41.54 gm) was normal. There was patchy interstitial fibrosis with myocyte disarray and hypertrophy (Fig. 4). An excess of adipose tissue was seen with an oil red-O stain on the liver, which was in keeping with a probable metabolic disorder. Electron microscopy demonstrated increased connective tissue between myocytes, which appeared thinner than expected with diminished contractile apparatus. Mitochondrial numbers were noted to be increased, focally enlarged, and their cytoplasm filled with transverse cristae. Dilated cardiomyopathy with subendocardial fibroelastosis was demonstrated. Barth syndrome was thought to be the underlying cardiac pathology, as this entity is an infantile-onset, X-linked recessive mitochondrial disorder, frequently presenting with dilated cardiomyopathy, primarily affecting males and females due to variants in TAZ encoding for the cardiolipin transacylase tafazzin. It has been proposed that the increase in the number of mitochondria seen in Barth Syndrome is a compensatory mechanism that prevents a decrease of ATP synthesis [11] (Fig. 5).
A male infant born at term but small for gestational age had cyanotic episodes 12 h into life, poor feeding, and dyspnea. Respiratory function deteriorated, and cardiac arrest ensued. Autopsy showed an enlarged heart (52.9 g, expected 30 + / − 7 g) with a globular morphology. The right ventricle was dilated and hypertrophic. On histology, there were prominent myocyte hypertrophy, frequent mitoses including some giant forms, and subendocardial fibroelastosis in both the right and left ventricles. Findings were of mitogenic cardiomyopathy; Fig. 4 depicts this extremely rare entity only recently described in the literature [12].
Fig. 4
a Myocyte hypertrophy and frequent mitosis characterizes mitogenic cardiomyopathy (H&E × 200); b Ki67 immunostain highlighted frequent mitotic figures (arrows) (× 200)
Fig. 5
Electron microscopy of the myocardium showed moderate increased number of mitochondria in cardiac myocytes. Some are also enlarged (2.4 μm in diameter, white arrow), and others are filled with transverse cristae (grey arrow) (magnification 30,000 ×). Courtesy of Mr Bart Wagner, Head of Electron Microscopy at Sheffield Teaching NHS FT
Vascular lesionsAlthough these conditions are relatively rare in children, we observed 14 cases. The most frequent condition was the rupture of a clinically undiagnosed thoracoabdominal aortic aneurysm (4 cases). Some of the unusual cases are summarized below:
A 16-year-old female with a history of chest pain, asthma, and short intervals of central cyanosis when exposed to cold weather died suddenly. At PM, an anomalous coronary artery origin was noted, with the right coronary artery arising from the edge of the left coronary sinus and traversing the soft tissue plane between the pulmonary artery and aorta. The heart weight was normal (210 gm).
A 17-year-old male who died suddenly and unexpectedly had a thoracic aortic aneurysm due to an undiagnosed connective tissue disorder (suspected to be Loeys-Dietz syndrome). The weight of the heart (345 gm) was increased (expected for 60 kg male is 140–326 gm).
A 3-month-old female infant with a history of dyspnea and vomiting died from arterial calcification of infancy and subsequent papillary muscle rupture and acute myocardial infarction. The heart’s weight was increased (43 gm, expected 23.1–27.9 gm). Histology demonstrated subintimal fibrosis and calcification of the tunica media of numerous arteries within the heart, lungs, pancreas, periadrenal area, kidneys, thymus, and thyroid gland consistent with vascular calcification of infancy (Fig. 6a–d).
Other vascular processes seen included a case of fibromuscular dysplasia of the atrioventricular nodal artery in a white female infant with a history of ventricular septal defect, large patent ductus arteriosus (PDA) (3.5 mm in diameter), patent foramen ovale (PFO), and infantile respiratory distress syndrome (respiratory syncytial virus was isolated from the lungs and airway). She had several episodes of cardiac arrhythmias, cyanosis, and cardiac arrest. Autopsy findings included a muscular ventricular septal defect (VSD) of approximately 2 mm, and histopathology demonstrated normal myocardial and conduction system architecture, but focal and severe fibromuscular proliferation with near total luminal occlusion of the atrioventricular nodal artery. The PDA, PFO, and muscular VSD of approximately 2 mm diameter were confirmed at post-mortem. Fibromuscular dysplasia of intramyocardial arteries has been rarely described in the literature [13].
Many rarely described entities were identified in our study including long QT syndrome. One was a 3.5 years old female who developed sudden dyspnea over a period of 30 min and collapsed. She had a history of developmental delay, severe hearing impairment, anemia, and consanguineous parents. Macroscopic examination was unremarkable. The heart’s weight was normal (82.86 gm). The morphology of the sinoatrial nodes revealed abundant nerve fibers, an artery and fusiform fibers embedded in fibrous connective tissue. Due to her deafness, Jervell-Lange-Nielsen syndrome was suspected. This was confirmed by post-mortem mutational analysis, which revealed the presence of a nonsense mutation of KCNE1 at 21q22.12, thus confirming the diagnosis of Jervell-Lange-Nielsen syndrome.
Fig. 6
A female infant with a history of dyspnoea and vomiting, whose post-mortem showed Idiopathic Arterial Calcification of Infancy, which caused secondary papillary muscle rupture and acute myocardial infarction: a coronary artery calcification (H&E × 200); b extensive myocardial infarction of the left ventricle; c calcification arteries in the thyroid (H&E × 200); and d pancreas among many other organs (H&E × 200)
Cardiac malformations/complications post-surgeryUndiagnosed cardiac malformations or complications after surgical repair were the most common etiology of sudden unexpected death in our cohort (51 cases). In 21/51 cases, deaths occurred after repaired malformations (Table 1). Atrioventricular septal defects, transposition of great vessels, and hypoplastic left heart were the most common conditions in this group. The more interesting cases are summarized below:
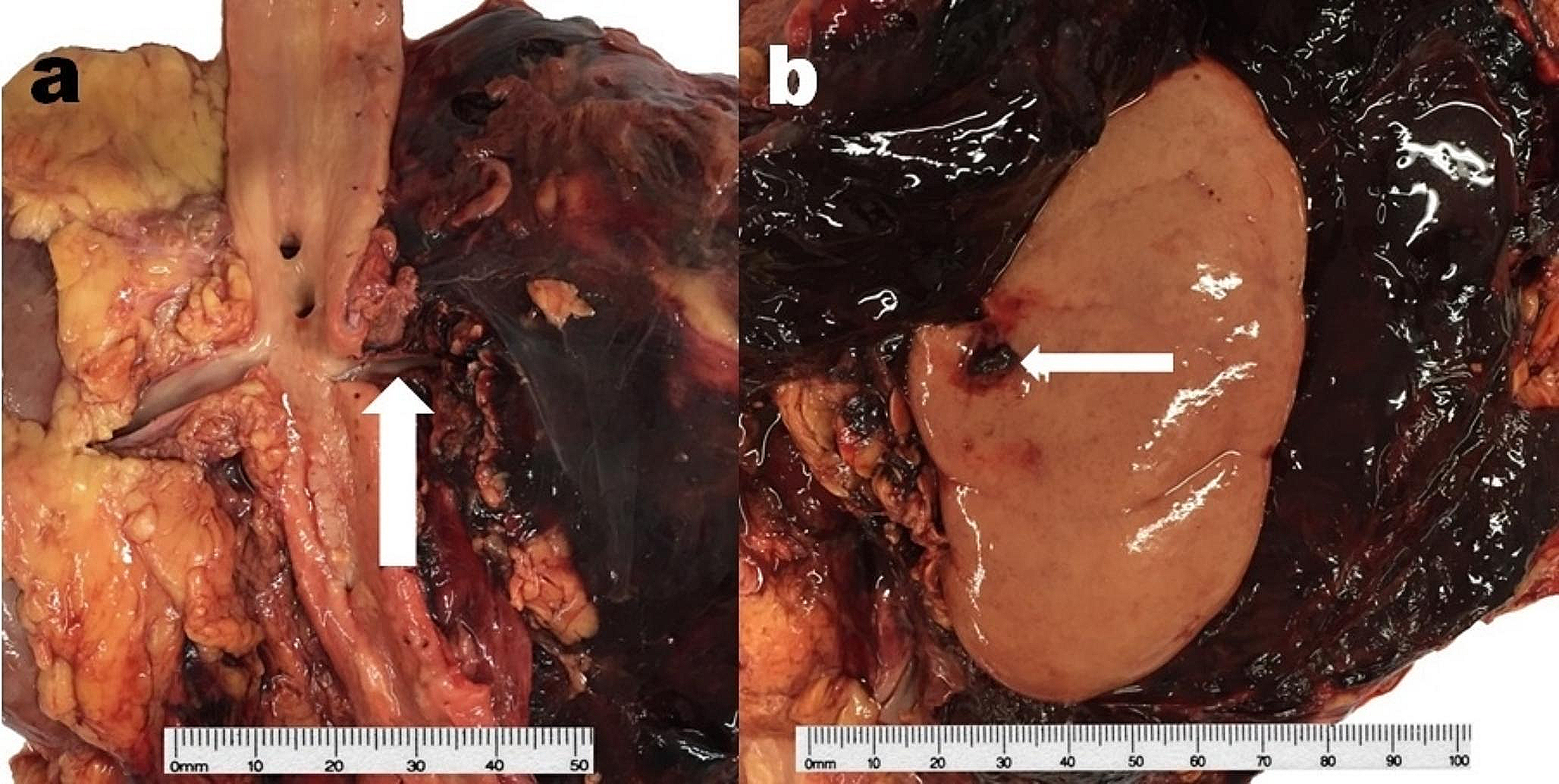
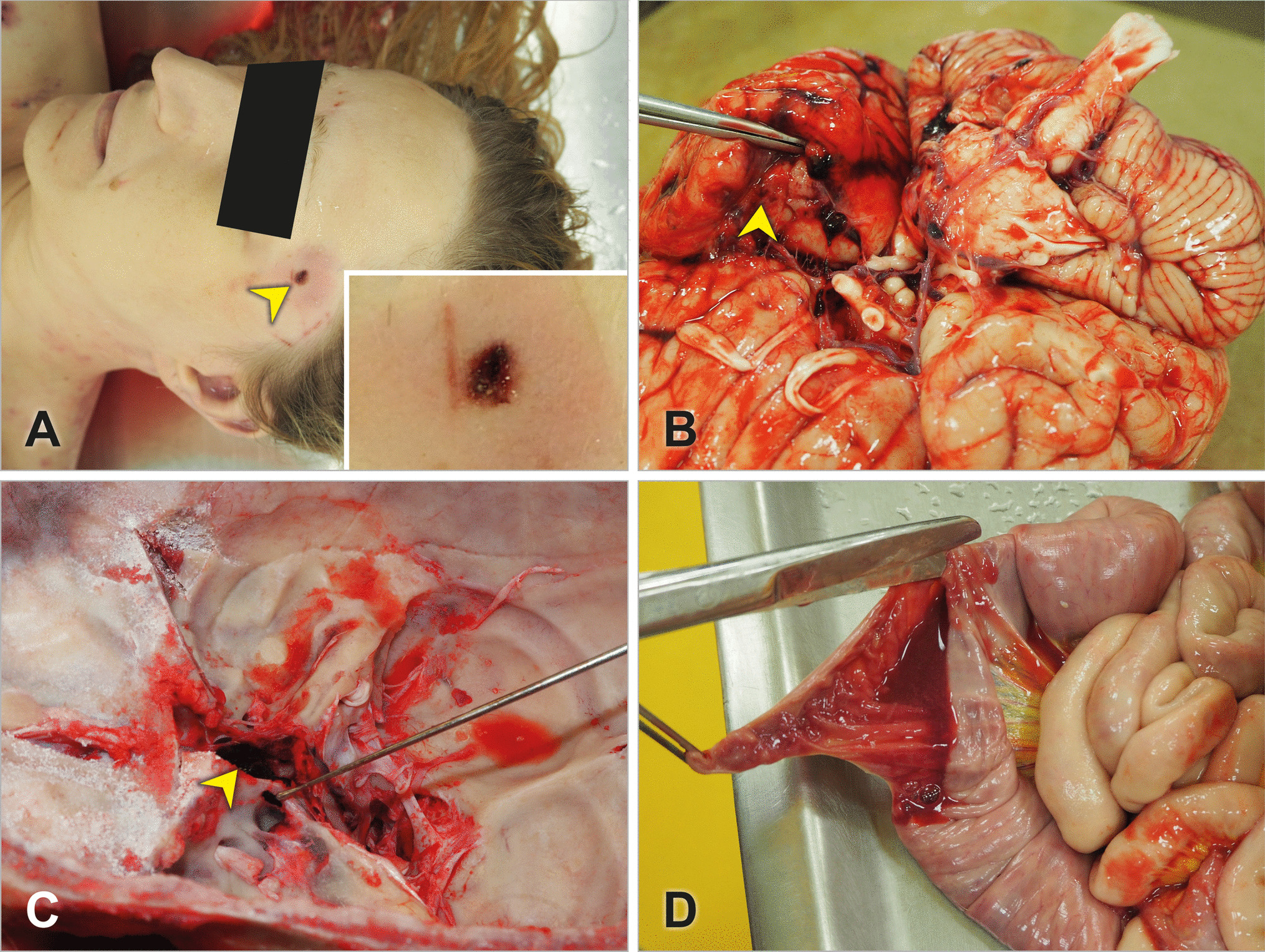
An 18 months old female toddler twin with a history of prematurity (corrected age: 14 months) and a large PDA underwent a transcatheter closure procedure. Three days later, she developed vomiting and reduced Glasgow Coma Scale (GCS). PM examination revealed the cause of death to be related to migration of the PDA closure device, which was found in the abdominal aorta. The device was surrounded by a thrombus, occluding the aorta. The heart’s weight was normal (63 gm) (Fig. 7a–c).
A 12 years old female with a history of cyanotic and complex congenital heart disease including pulmonary atresia, ventricular septal defect, multiple pulmonary aortopulmonary collateral arteries (MACPCAs), hypoplastic central pulmonary arteries with peripheral branching stenosis, and Alagille syndrome with early portal hypertension underwent cardiac catheterization, stenting, and ballooning of the right pulmonary artery. She presented 1 year later with a viral upper respiratory tract infection, a Staphylococcus aureus bacteremia, and subsequent bacterial endocarditis involving her right ventricular pulmonary artery conduit. She underwent surgery to replace the infected conduit with a Hancock pericardial xenograft valve; however, empyema, which was drained, was noted in the mediastinum. She had a poor postoperative recovery requiring intubation, ventilation, and inotropic support before suffering cardiac arrest. The autopsy revealed the repaired pulmonary trunk with the Hancock xenograft and a repaired peri-membranous ventricular septal defect. The heart’s weight was increased (350 gm, expected: 124 gm). Pulmonary arteries were occluded by thrombi, and there was aneurysmal dilatation extending into the hila of both lungs. Histology revealed occlusive thrombi and aneurysmal dilatation of both pulmonary arteries and a stent surrounded by bacterial colonies of P. putrida and infective endocarditis with dense neutrophilic infiltration extending up from the graft.
Fig. 7
A female toddler with a large patent ductus arteriosus (PDA) underwent a transcatheter closure procedure. Three days later, she developed vomiting and reduced Glasgow Coma Scale (GCS). Cause of death was attributed to the migration of the PDA closure device, which was found in the abdominal aorta. a the PDA device was visualized on the X-ray conducted as part of the post-mortem at the level of T12 (arrow); b posterior view of the thoracoabdominal eviscerated block, demonstrating the migrated device in the abdominal aorta (note it below the diaphragm and just above both kidneys); c the PDA migrated device was surrounded by a fresh mural thrombus (H&E × 100)
留言 (0)