
記住我










Diabetic retinopathy (DR) is a leading cause of vision loss in working-age adults. Despite an established standard of care for advanced forms of DR, some patients continue to lose vision after treatment. This may be due to the development of diabetic macular ischemia (DMI), which has no approved treatment. Neuropilin-1 (Nrp-1) is a coreceptor with two ligand-binding domains, with semaphorin-3A (Sema3A) binding to the A-domain and vascular endothelial growth factor-A (VEGF-A) binding to the B-domain. Sema3A directs a subset of neuronal growth cones as well as blood vessel growth by repulsion; when bound to Nrp-1, VEGF-A mediates vascular permeability and angiogenesis. Modulating Nrp-1 could therefore address multiple complications arising from DR, such as diabetic macular edema (DME) and DMI. BI-Y is a monoclonal antibody that binds to the Nrp-1 A-domain, antagonizing the effects of the ligand Sema3A and inhibiting VEGF-A–induced vascular permeability. This series of in vitro and in vivo studies examined the binding kinetics of BI-Y to Nrp-1 with and without VEGF-A165, the effect of BI-Y on Sema3A-induced cytoskeletal collapse, the effect of BI-Y on VEGF- A165–induced angiogenesis, neovascularization, cell integrity loss and permeability, and retinal revascularization. The data show that BI-Y binds to Nrp-1 and inhibits Sema3A-induced cytoskeletal collapse in vitro, may enhance revascularization of ischemic areas in an oxygen-induced retinopathy mouse model, and prevents VEGF-A–induced retinal hyperpermeability in rats. However, BI-Y does not interfere with VEGF-A–dependent choroidal neovascularization. These results support further investigation of BI-Y as a potential treatment for DMI and DME.
SIGNIFICANCE STATEMENT Diabetic macular ischemia (DMI) is a complication of diabetic retinopathy (DR) with no approved pharmacological treatment. Diabetic macular edema (DME) commonly co-occurs with DMI in patients with DR. This series of preclinical studies in mouse and rat models shows that neuropilin-1 antagonist BI-Y may enhance the revascularization of ischemic areas and prevents vascular endothelial growth factor-A (VEGF-A)–induced retinal hyperpermeability without affecting VEGF-A–dependent choroidal neovascularization; thus, BI-Y may be of interest as a potential treatment for patients with DR.
IntroductionDiabetic retinopathy (DR) is a leading cause of vision loss in working-age adults, with an estimated global prevalence of 27%–35% (Lee et al., 2015; Simó-Servat et al., 2019; Thomas et al., 2019). Diabetic macular edema (DME) is considered responsible for most of the vision loss associated with DR; approximately one-third of patients with DR have comorbid DME (Lee et al., 2015). Although anti–vascular endothelial growth factor-A (anti–VEGF-A) therapies are the established standard of care for DME, some patients continue to lose vision after otherwise successful treatment, potentially due to the development of diabetic macular ischemia (DMI) (Bressler et al., 2022). Depending on imaging modality, disease severity, and definition, up to 77% of patients with DR are identified as having DMI (Sim et al., 2013). Although DMI currently lacks a formal definition, its key features comprise enlargement of the foveal avascular zone, reduced mean retinal arteriolar diameter, and density loss of the macular capillary network (Conrath et al., 2005; Liew et al., 2015; Garcia et al., 2016).
Ischemia-related damage to the retina and surrounding vasculature can form a vicious cycle wherein retinal capillary damage leads to retinal damage and retinal damage promotes retinal capillary damage (Usman 2018). The neuroretinal damage resulting from DMI can lead to progressive vision loss (Sim et al., 2013; Sorour et al., 2018; Usman 2018) (e.g., enlargement of the foveal avascular zone correlates with reduced visual acuity) (Balaratnasingam et al., 2016). Despite the proven clinical burden of vision loss, no therapies are currently approved for the management or treatment of patients with DMI (Sim et al., 2013; Usman, 2018). In addition, DMI may negatively affect the efficacy of anti–VEGF-A therapies, which are commonly used to treat other complications of DR (Manousaridis and Talks, 2012), highlighting the unmet clinical need for DMI-specific treatments.
In physiologic angiogenesis, endothelial tip cells guide new vessels by extending filopodia that contain a high density of growth factor receptors; this enables new vessels to detect growth-factor gradients and thus migrate by vasoattraction or vasorepulsion (Gerhardt et al., 2003; Ochsenbein et al., 2016). In proliferative retinopathies, this process is disrupted (Joyal et al., 2011). Initial degeneration of the retinal microvasculature is followed by hypervascularization as the ischemic retina attempts to restore metabolic equilibrium (Joyal et al., 2011). The new vessels fail to propagate toward ischemic regions and instead grow into the vitreous, suggesting pathologic misdirection by vasorepulsive cues (Joyal et al., 2011). Breakdown of the inner blood–retina barrier, leading to disrupted vascular permeability and potential for retinal edema, is also associated with complications of DR such as DME (Antonetti et al., 1999). Normalizing vascular function in DR through modulation of vasoattraction and vasorepulsion as well as vascular permeability may restore areas of ischemia and reduce edema, breaking the cycle of damage.
One receptor characterized as mediating both vascular guidance and permeability is Nrp-1. Nrp-1 signaling prompts endothelial cells to heed attractive and repulsive guidance cues, thus regulating the direction of vessel growth (Carmeliet and Jain, 2011; Ochsenbein et al., 2016; Roth et al., 2016). Nrp-1 has two extracellular domains, A and B, that respectively bind semaphorin-3 proteins and VEGF proteins, including semaphorin-3A (Sema3A) and VEGF-A (Appleton et al., 2007; Roth et al., 2016).
Sema3A is a guidance molecule that directs vessel growth by vasorepulsion (Moret et al., 2007; Cerani et al., 2012; Iragavarapu-Charyulu et al., 2020). Under hypoxic conditions, several retinal cells, such as retinal ganglion cells, secrete Sema3A (Joyal et al., 2011). By binding to Nrp-1 and plexin-A, Sema3A then induces a cytoskeletal collapse in the filopodia of endothelial tip cells, thus leading to vasorepulsion of the newly forming retinal vessels (Joyal et al., 2011). Modulation of Nrp-1 activity may alter the vasorepulsive actions of Sema3A, thereby improving ischemia.
VEGF-A is upregulated in various retinopathies, including DR, and alters both vascular permeability and capillary perfusion (Pe’er et al., 1996; Hammes et al., 1998; Miyamoto et al., 2000); this can lead to edema (i.e., DME) (Romero-Aroca et al., 2016). As Nrp-1 is a coreceptor for VEGF-A and regulates vascular permeability signaling (Domingues and Fantin, 2021), modulating Nrp-1 may improve retinal edema.
The role of Nrp-1 in neovascularization and vascular permeability has been explored using oxygen-induced retinopathy (OIR) mouse models. In the OIR model, mice with Nrp-1 hypomorphism (partial loss of function) show reduced angiogenesis compared with wild-type mice (Fantin et al., 2014). In addition, OIR mice lacking endothelial Nrp-1 have reduced leakage into the retina (Fernández-Robredo et al., 2017).
BI-Y is a monoclonal IgG1 type antibody for intravitreal injection that binds to the A-domain of Nrp-1 and antagonizes its biologic effects. The in vitro and in vivo studies presented here examined: 1) the binding of BI-Y to Nrp-1 with and without the presence of VEGF-A165, 2) the effect of BI-Y on in vitro Sema3A-induced cytoskeletal collapse, and revascularization in an OIR mouse model, and 3) the effect of BI-Y on VEGF-A–induced in vitro angiogenesis, neovascularization in a rat model of laser-induced choroidal neovascularization, and in vitro cell integrity loss and permeability in a rat model of VEGF-A–induced retinal hyperpermeability.
Materials and MethodsAll studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals, as adopted and promulgated by National Institutes of Health, and were reviewed by a Federal Ethics Committee and approved by government authorities in Germany.
BI-Y and Nrp-1Binding Kinetics of BI-Y to Nrp-1Binding kinetics were measured via surface plasmon resonance using a ProteOn XPR36 (Bio-Rad Laboratories, Hercules, CA). BI-Y (0.6 μg/ml) was captured on a Protein A/G (Thermo Scientific, Waltham, MA) surface vertically for 300 seconds at a flow rate of 30 μl/min. Phosphate-buffered saline (PBS)-Tween20-EDTA (Bio-Rad Laboratories) was injected horizontally for 60 seconds at a flow rate of 100 μl/min and with a dissociation time of 120 seconds. Human, cynomolgus monkey, mouse, or rat Nrp-1 was injected horizontally over the captured BI-Y for 300 seconds at a flow rate of 30 μl/min and with a dissociation time of 1800 seconds at concentrations of 0, 6.25, 12.5, 25, 50, and 100 nM.
The interspot (interactions with sensor surface) and blank PBS-Tween20-EDTA were subtracted from the raw data. Sensorgrams were then fitted to 1:1 Langmuir binding to provide on-rate (ka), off-rate (kd) and affinity (KD) values.
BI-Y and Sema3AThe Effect of BI-Y on Sema3A-Induced Cytoskeletal CollapseCells were seeded in Cells System Corporation (CSC, Kirkland, WA) complete medium at a seeding density of 20,000 cells per well and then incubated overnight under normal growth conditions. Concentration–response curves of recombinant human Sema3A in the absence and presence of 25, 62.5, 125, and 250 pM BI-Y were prepared; the cell index was measured using the xCELLigence device (Agilent Technologies, Santa Clara, CA). Individual experiments were performed in triplicate for Sema3A in the absence of BI-Y and in duplicate for Sema3A in the presence of BI-Y. For analysis (calculated 5 hours poststimulation), the cell index was normalized to the time point prior to the addition of substances. Potency was determined by the ability of BI-Y to shift the concentration–response curve [i.e., the 50% inhibitory concentration (IC50)] of Sema3A using Prism version 7.0 (GraphPad Software, CA) Gaddum Schild IC50 shift equations and was calculated from the pA2 value as potency = 10−x, where x is the pA2 value.
The Effect of BI-Y on the Number of Tip Cells at the Avascular Front, Revascularization of the Central Avascular Area, and Neovascularization in the Vitreous in an OIR Mouse ModelThe animals used were the pups of C57BL/6JRj mice (Janvier Laboratories, Le Genest-Saint-Isle, France). To induce OIR, at postnatal day (P) 7, mice were placed into oxygen chambers (BioSpherix, Parish, NY) in which the oxygen content was increased to 75% over 30 minutes; they remained in these chambers until P12. At the end of each day, the mice underwent a short period of normoxia; oxygen content was decreased to room conditions over a 2-hour period, held at room conditions for 30 minutes, and then returned to 75% over a 30-minute period. After return to normoxia at P12, the mice received a single 10-μg intravitreal injection of BI-Y in each eye, IgG anti-trinitrophenol control antibody, or aflibercept (Komtur Pharmaceuticals, Freiburg im Breisgau, Germany) at a volume of 0.5 μl under isoflurane anesthesia. At P17, the mice were killed by cervical dislocation under isoflurane anesthesia, and the eyes were enucleated. The left eye of each animal was fixed in paraformaldehyde (4%) for 1 hour at 4°C and then transferred to PBS. The retinae were isolated, stained with fluorescein isothiocyanate–labeled isolectin B4 (L9381; Sigma-Aldrich, St. Louis, MO), and incubated overnight at 4°C. Each retina was transferred to a glass slide and cut four times to achieve a flat cloverleaf-like structure.
The samples were excited at a wavelength of 488 nm with an LSM 700 confocal laser scanning microscope (Carl Zeiss AG, Oberkochen, Germany), and images of the retinal flat mounts were obtained. Total retinal area and avascular retinal area were measured using Zeiss ZEN software (version ZEN 3.1; blue edition). Tip cells located at the avascular front were manually counted by a masked observer, and the length of the avascular front was measured using ImageJ (Wayne Rasband, National Institutes of Health, version 1.50e). The right eye of each animal was fixed in 4% paraformaldehyde at 4°C for 24 hours and embedded in paraffin. Sagittal sections (6 μm) extending in both directions from the optic nerve were prepared. Each third section was stained with the periodic acid-Schiff technique. Intravitreal neovascularization was then assessed by counting the preretinal nuclei, and the mean count of 10 sections was calculated per eye.
BI-Y and VEGF-ASimultaneous Binding of BI-Y and VEGF-A165 to Nrp-1The competitive binding of human VEGF-A165 and BI-Y to human Nrp-1 was investigated using the OctetRED96 system (Octet Software Version 9.0.0.4; ForteBio, Fremont, CA). Streptavidin sensor tips (ForteBio) were immersed in a biotinylated human VEGF-A165 solution (10 μg/ml) prepared in kinetic buffer (ForteBio) for 2 minutes. Human Nrp-1 (100 nM) prepared in kinetic buffer (ForteBio) was then captured via the human VEGF-A165 for 10 minutes. Finally, the sensors were dipped into two concentrations of BI-Y (100 nM and 400 nM) for 10 minutes. Data from active sensors were compared with those from three control conditions: no human VEGF-A165 capture, no human Nrp-1, and no antibody.
The Effect of BI-Y on In Vitro VEGF-A–Induced Angiogenesis(a) The effect of BI-Y on prevention of VEGF-A–induced endothelial network formationNormal human dermal fibroblasts (NHDFs; Lonza Group, Basel, Switzerland) were seeded in a mixture of fibroblast growth medium 2 and endothelial growth medium (EGM; PromoCell, Heidelberg, Germany) in equal parts, at a density of 25,000 cells per well and cultured for 1 week under normal growth conditions.
The medium was removed from the NHDFs (Lonza Group), and human umbilical vein endothelial cells (HUVECs; PromoCell) were seeded at a density of 5000 cells per well in 1/10 EGM/endothelial basal medium (EBM) mixture (PromoCell) on top of the NHDFs. HUVECs were allowed to attach in the incubator for 4 hours. Then the medium was removed, and the cells were stimulated with 10 ng/ml recombinant human VEGF-A (R&D Systems, Minneapolis, MN) plus 50 μl of solution containing either BI-Y or aflibercept (final concentrations: 1.6−8, 5.06−9, 1.6−9, 5.06−10, 1.6−10, 5.06−11, and 1.6−11 M). The controls were 1/10 EGM/EBM mixture alone and human VEGF-A (10 ng/ml) without BI-Y. Each condition was performed six times per individual experiment.
The cells were stained with 50 μl per well of 1 μg/ml anti-CD31 in Dulbecco’s phosphate-buffered saline containing 1% bovine serum albumin and stored in 200 μl per well PBS at 4°C until ready for microscopy. The plates were imaged using the Opera Phenix (PerkinElmer, Waltham, MA) screening system, and the 488-positive network area per well was calculated using Harmony (version 4.9; PerkinElmer).
The data were then analyzed relative to the VEGF-A effect, which was calculated as mean VEGF-A–induced network area minus mean basal network area. IC50 values were calculated from each respective curve using Prism version 7.0 (GraphPad Software) curve fitting. A geometric mean of the IC50 values of two experiments was calculated using Microsoft Excel (2016). The maximal efficacy was calculated as the mean of two experiments at the highest antibody concentrations as percent inhibition of the VEGF-A–induced network area.
(b) The effect of BI-Y on VEGF-A–induced endothelial proliferationHuman retinal microvascular endothelial cells (HRMECs; CSC) were seeded at a density of 3000 cells per well in EGM (PromoCell) and allowed to attach in the incubator for approximately 17 hours. Then the growth medium was replaced by endothelial basal medium (EBM) containing 2% fetal calf serum, and cells were incubated for 7.5–8.5 hours under normal growth conditions in the incubator. A stimulation medium containing recombinant human VEGF-A (R&D Systems) at a final concentration of 10 ng/ml plus either aflibercept (Komtur Pharmaceuticals, Freiburg im Breisgau, Germany) or BI-Y (final concentrations: 1.6 nM, 4 nM, 1 nM, 250 pM, 62.5 pM, 15.6 pM), was preincubated for 1 hour then added to the cells.
Cells were cultured under normal culture conditions for live cell imaging (Incucyte; Sartorius AG, Gottingen, Germany). Four phase-contrast images per well were taken every 4 hours for 96 hours. Each condition was performed six times per individual experiment. For analysis, resulting data were normalized for each individual well to the time point prior to stimulation. Growth curves over a time span of 96 hours were depicted, and the area under the curve (AUC) was calculated using Prism version 7.0 (GraphPad Software). For the evaluation of efficacy and potency, the mean AUC of unstimulated cells (basal) was subtracted from the individual AUC values. Concentration–response curves were depicted using Prism version 7.0 (GraphPad Software) curve fitting, and IC50 values were calculated from each respective curve. A geometric mean of the IC50 values was calculated using Microsoft Excel (2016). The efficacy was calculated at the highest antibody concentrations as percent inhibition of the VEGF-A–induced network area.
The Effect of BI-Y on In Vivo VEGF-A–Dependent NeovascularizationBrown Norway rats (male, weight range 160–180 g; obtained from Charles River Laboratories, Sulzfeld, Germany) underwent laser photocoagulation under anesthesia to induce choroidal neovascularization. Laser treatment was performed with a green Argon laser (532 nm, Merilas; Meridian, Thun, Switzerland) using a Micron IV system (Phoenix Research Laboratories, Pleasanton, CA). The diameter of the laser beam was matched with the diameter of the optic nerve, and a 150-millisecond, 400-mW laser pulse was used to generate four lesions per eye. The rats then received two intravitreal injections of either an IgG control antibody (109 μg/eye), aflibercept (200 μg per eye), or BI-Y in each eye of either a low dose (54.5 μg per eye) or a high dose (109 μg per eye). The first injection was performed immediately after laser treatment (day 1), and the second injection was performed on day 8.
Fourteen days after laser treatment, the rats were killed by cervical dislocation under anesthesia, and the eyes were enucleated. The eye cup (consisting of the retinal pigment epithelium, choroidea, and sclera) was fixed in paraformaldehyde (4%) for 1 hour at 4°C and then transferred to PBS containing 0.1% Triton X-100 (Sigma-Aldrich) for 1 hour at 4°C. The eye cup was stained overnight with fluorescein-5-isothiocyanate–labeled isolectin B4 (10 μg/ml in saline; Sigma Aldrich) and then transferred to a glass slide and cut four times to achieve a flat cloverleaf-like structure; flat mounts were stored at 4°C in the dark until analysis at a wavelength of 488 nm with an LSM 700 confocal laser scanning microscope (gain 650, laser strength 2%; Carl Zeiss AG). Measurement of the lesion size was done with Zeiss Zen Blue software (version ZEN 3.1; blue edition).
The Effect of BI-Y on VEGF-A–Induced Cell Integrity LossCell integrity was determined as a reduction in the ‘cell index,’ a measure of cellular impedance using the xCELLigence system (Agilent Technologies). For data analysis, the cell index was normalized to the time point prior to the addition of substances. Cells were seeded in CSC complete medium at a density of 20,000 cells per well and incubated overnight under normal growth conditions inside the xCELLigence device.
The growth medium was replaced by CSC basal medium supplemented with 0.5% bovine serum albumin; cells were then incubated in the xCELLigence device for 3 hours. Subsequently, cells were stimulated with recombinant human VEGF-A (20 ng/ml; R&D Systems) plus BI-Y (final concentrations: 1−7, 2−8, 4−9, 8−10, 1.6−10, 3.2−11, and 6.4−12 M). Basal medium with either 0.5% bovine serum albumin or VEGF-A without antibodies served as controls.
For analysis, the cell index was normalized to the time point prior to the addition of substances. The results were calculated at the peak drop in cellular resistance (22–24 minutes after the cells were stimulated). IC50 values were calculated from each respective curve with XLfit (IDBS, Alameda, CA) curve fitting on concentration-response results. A geometric mean value was calculated using Microsoft Excel (2016).
The Effect of BI-Y on VEGF-A–Induced Permeability in the RetinaBrown Norway rats (male, weight range 160–180 g; Charles River Laboratories, Sulzfeld, Germany) received a single 2.5-μl intravitreal injection of BI-Y in each eye (38.75 μg), IgG control antibody (38.75 μg), or aflibercept (100 μg; Komtur Pharmaceuticals) while under isoflurane anesthesia. After 15 minutes and within the same anesthesia, the rats were intravitreally injected at the same site with 100 ng or 250 ng of recombinant human VEGF-A (R&D Systems, Minneapolis, MN) or PBS (2.5 μl).
For visualization of retinal hyperpermeability, 24 hours later Evans Blue dye (Sigma Aldrich, Germany; 45 mg/ml in 0.9% saline) was intravenously injected at 1 ml/kg under isoflurane anesthesia. The animals were killed by cervical dislocation 30 minutes after the Evans Blue infusion; the eyes were enucleated, and the eye cup was fixed in paraformaldehyde (4%) for 1 hour at 4°C and then transferred to PBS overnight at 4°C. The retinae were separated from the outer segments (sclera and choroidea) and transferred to a glass slide and cut four times to achieve a flat cloverleaf-like structure. The samples were excited at a wavelength of 639 nm, and emission of Evans Blue at 669 nm was recorded with an LSM 700 confocal laser scanning microscope (Carl Zeiss AG). Analysis of fluorescence intensity sum was done with the program ImageJ (Wayne Rasband, National Institutes of Health, version 1.50e).
ResultsBI-Y and Nrp-1Binding Characteristics of BI-Y to Nrp-1 in Surface Plasmon Resonance AssaysBI-Y bound to human Nrp-1 with a ka of 2.29 × 105 1/Ms, a kd of 2.54 × 10−3 1/s, and a KD of 1.11 × 10−8 M (for full results, see Table 1).
TABLE 1The binding characteristics of BI-Y to human, cynomolgus monkey, mouse, and rat Nrp-1
BI-Y and Sema3ABI-Y Prevents Sema3A-Induced Cytoskeletal Collapse in Primary HRMEC AssaysBI-Y prevented Sema3A-induced cytoskeletal collapse in primary HRMECs with a potency of 98 pM (pA2 value of 10.007; example curve shift shown in Fig. 1).
 Fig. 1.
Fig. 1. Determination of the pA2 values for BI-Y to prevent a Sema3A-induced cytoskeletal collapse in HRMEC in one representative IC50 shift experiment. Data points are presented as the mean ± S.E.M.
BI-Y Increases Tip Cell Density and Reduces Avascular Area in an OIR Mouse ModelSema3A was upregulated in retinal lysates of OIR mouse pups compared with age-matched control animals (Supplemental Fig. 1). Tip cell density (i.e., the number of tip cells at the avascular front in retinal flat mounts) increased by 48% (P < 0.001) in eyes treated with BI-Y relative to eyes injected with an IgG control antibody; eyes treated with aflibercept showed a reduction of 5% (not statistically significant) in tip cell density (Fig. 2A). Representative images of tip cells have been published in Zippel et al. (2022). The avascular area in eyes treated with BI-Y decreased by 35% relative to eyes injected with an IgG control antibody (not statistically significant); eyes treated with aflibercept showed an increase in avascular area of 24% (not statistically significant; Fig. 2B). Representative images of retinal flat mounts showing the avascular area are provided in Supplemental Fig. 3. Compared with the aflibercept group, the avascular area in the BI-Y group was significantly lower (47%, P < 0.05). Finally, the number of preretinal tufts decreased by 17% (not statistically significant) in eyes treated with BI-Y relative to eyes injected with an IgG control antibody and decreased by 61% (P < 0.01) in eyes treated with aflibercept (Fig. 2C). Tip cell density was found to significantly negatively correlate with avascular area (r = −0.6142, P < 0.0001; Fig. 2D).
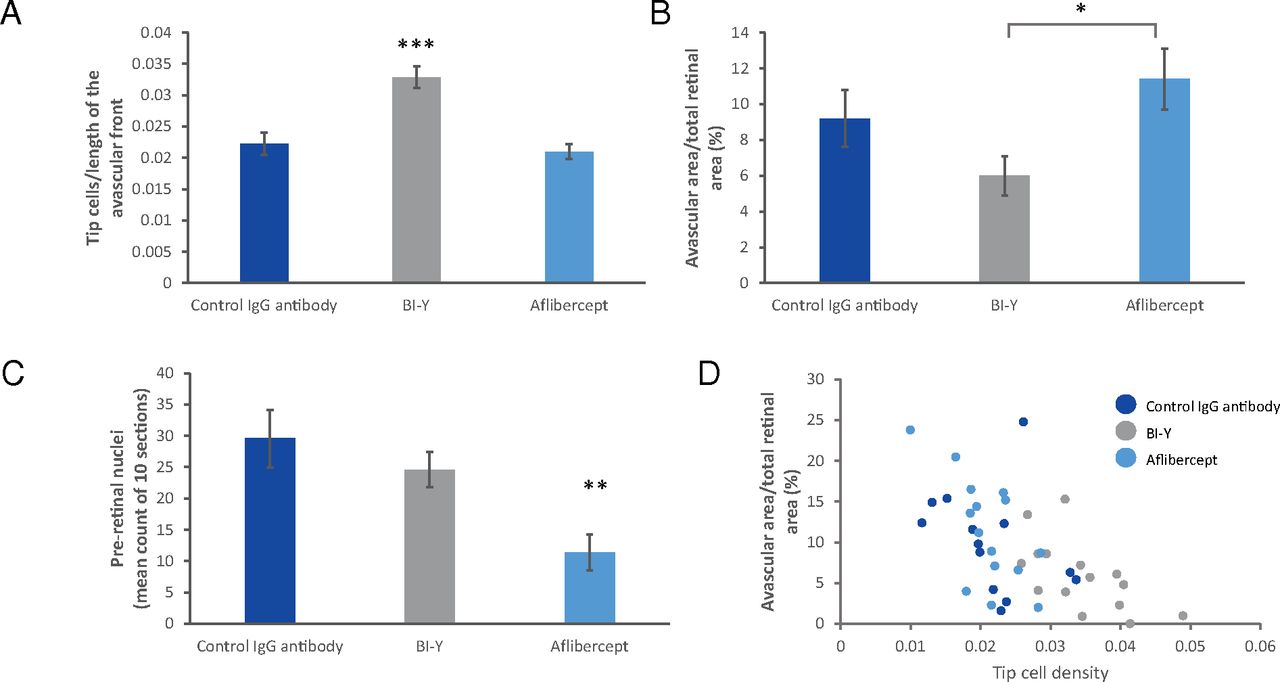 Fig. 2.
Fig. 2. BI-Y treatment in an oxygen-induced retinopathy mouse model at postnatal day 17. (A) Tip cell density in retinal flat mounts of mouse pups. Data are mean ± S.E.M., n = 14–18. Statistical analysis was performed by a one-way ANOVA analysis with Dunnett’s multiple comparisons test. (B) Avascular area in retinal flat mounts of mouse pups. Data are mean ± S.E.M., n = 15. Statistical analysis was performed by unpaired two-sided t test. (C) Preretinal nuclei in enucleated eyes of mouse pups. Data are mean ± S.E.M., n = 17. Statistical analysis was performed by unpaired two-sided t test vs. control. (D) Correlation between tip cell density and avascular area in retinal flat mounts of mouse pups. Data points are the values for each individual eye, n = 42. *P < 0.05; **P < 0.01; ***P < 0.001.
BI-Y and VEGF-ABI-Y and Human VEGF-A165 Simultaneously Bind to Human Nrp-1Human VEGF-A165 and BI-Y simultaneously bound to Nrp-1 without competition between binding partners (Fig. 3A). Control experiments examining human VEGF-A165 alone, human Nrp-1 alone, and BI-Y alone showed no recorded response, validating the notion that the observed binding signals were specific to the interaction between human VEGF-A165/BI-Y and human Nrp-1.
 Fig. 3.
Fig. 3. (A) The binding of BI-Y to Nrp-1 in the presence of VEGF-A (data from one representative experiment). Dissociation after human Nrp-1 binding to human VEGF-A165 without BI-Y is shown in blue. A second association after the addition of BI-Y is shown in gray and yellow. (B) The effect of BI-Y and aflibercept on VEGF-A–induced network formation. Data points from one example experiment are presented as the mean ± S.D. (C) The effect of BI-Y and aflibercept on VEGF-A–induced endothelial cell proliferation. Data points from one example experiment are presented as the mean AUC ± S.E.M, calculated using GraphPad Prism as the mean AUC of unstimulated cells subtracted from individual AUC values. For the individual growth curves, see Supplemental Fig. 2. (D) The effect of BI-Y and aflibercept on choroidal neovascular lesion area vs. a control IgG antibody in Brown Norway rats after ocular laser photocoagulation. Data are mean ± S.E.M., n = 30–39. Statistical analysis was done by unpaired two-sided t test (****P < 0.01). (E) The effect of BI-Y on VEGF-A–induced cell integrity loss in HRMECs. Data points from one example experiment are presented as the mean ± S.D. The orange line represents the mean of the basal control, and the blue line represents the mean of VEGF-A–treated cells without BI-Y. (F) Permeability in retinal flat mounts of Brown Norway rats with and without VEGF-A after treatment with BI-Y, aflibercept, or a control IgG antibody. Data are mean ± S.E.M., n = 8–12. Statistical analysis was done by unpaired two-sided t test (**P < 0.01).
BI-Y Demonstrates No Prominent Effect on VEGF-A–Induced In Vitro Angiogenesis in Functional AssaysBI-Y had no relevant effect on VEGF-A–induced endothelial network formation, whereas aflibercept resulted in a mean reduction of 80% with an IC50 of 0.09 nM (Fig. 3B). BI-Y did not prevent VEGF-A–induced endothelial cell proliferation (2.0% reduction in VEGF-A effect), whereas aflibercept was efficacious (86.4% reduction in VEGF-A effect, potency of 0.12 nM; Fig. 3C).
BI-Y Does Not Affect VEGF-A–Dependent Laser-Induced Choroidal Neovascularization in RatsBI-Y had no relevant effect on the size of the neovascular lesions (Fig. 3D). The mean lesion area was 144,671 μm2 for BI-Y (low dose) and 137,943 μm2 for BI-Y high dose, respectively, versus 142,505 μm2 for the IgG control antibody at day 15. Lesion area was decreased by 24% (P < 0.0001) versus control eyes after treatment with aflibercept (lesion area 107,595 μm2). Representative images of laser-induced lesions are shown in Supplemental Fig. 4.
BI-Y Partially Prevents VEGF-A–Induced Cell Integrity LossBI-Y partly prevented VEGF-A–induced cell integrity loss with a potency 0.97 nM (Fig. 3E).
BI-Y Prevents VEGF-A–Induced Retinal Hyperpermeability in a Rat ModelRetinal permeability increased by a mean of 121% (P < 0.01) after injection of VEGF-A (Fig. 3F). However, BI-Y prevented VEGF-A–induced retinal hyperpermeability in rats to the same extent as aflibercept: injection of either compound was found to completely inhibit VEGF-A–induced retinal hyperpermeability. Representative images of retinal flat mounts showing the extravasation of Evans Blue are given in Supplemental Fig. 5.
DiscussionNrp-1 is a receptor for Sema3A (Joyal et al., 2011), a molecule that inhibits angiogenesis by vasorepulsion (Moret et al., 2007; Cerani et al., 2012; Iragavarapu-Charyulu et al., 2020) and is secreted by retinal ganglion cells under hypoxic conditions (Joyal et al., 2011). Research using mice with Nrp-1 knockdown within the circulatory system has shown that Sema3A-mediated breakdown of inner blood–retinal barrier function is Nrp-1 dependent (Cerani et al., 2013). Nrp-1 also binds VEGF-A, a protein involved in multiple processes in the vasculature, including angiogenesis, cellular permeability, and vascular tone; mice with mutated Nrp-1 show reduced vascular permeability (Roth et al., 2016). Arrangement of the receptor tyrosine kinase VEGF receptor 2 (VEGFR2), the main VEGF-A receptor on endothelial cells, with additional proteins including Nrp-1, may lead to numerous different holoreceptor complexes. The composition of these complexes determines the downstream consequences and subsequent signaling pathways (Gu et al., 2003; Escalante and Zalpour, 2011; Shibuya, 2011; Pandey et al., 2018). Therefore, modulation of both Sema3A and VEGF-A via Nrp-1 could improve retinal pathologies associated with their dysfunction in humans, including ischemia (e.g., DMI) and edema (e.g., DME) (Thomas et al., 2021).
In this series of preclinical studies, we show that BI-Y potently binds to human Nrp-1 and is noncompetitive with human VEGF-A165. Furthermore, we demonstrate that BI-Y inhibits Sema3A-induced cytoskeletal collapse in isolated primary HRMECs in vitro, indicating that it could modulate vascular guidance. This conjecture is supported by our finding that, when compared with aflibercept, intravitreal administration of BI-Y into the eye of mice with OIR significantly reduces the size of the avascular area and significantly increases tip cell density at the avascular front. These results align with previous work demonstrating the central role of Sema3A and Nrp-1 in angiogenesis and endothelial leakage using mouse models (Fantin et al., 2014; Fernández-Robredo et al., 2017; Nakamura et al., 2022; Zippel et al., 2022). Nakamura et al. (2022) found that induction of vein occlusion significantly increases retinal Sema3A expression, and Zippel et al. (2022) showed that application of a Sema3A neutralizing antibody (referred to as BI-X) in an OIR resulted in a significant negative correlation between tip cell density and size of the avascular area and a trend for reduction of the avascular area (i.e., the effects of the two antibodies are qualitatively similar). Furthermore, reduced angiogenesis has been observed in mice with Nrp-1 hypomorphism, and OIR mice lacking endothelial Nrp-1 have reduced retinal edema when compared with wild-type mice (Fantin et al., 2014; Fernández-Robredo et al., 2017). Importantly, the negative correlation observed here between tip cell density and avascular area indicates that larger numbers of tip cells lead to new vessel growth in ischemic areas; therefore, increased tip cell density is necessary for revascularization of the avascular area.
The data presented here do not establish the specific VEGF-A mechanism or pathway that is impacted by BI-Y; however, it appears to have selective effects on the VEGF-A receptor complex. Although BI-Y had no relevant effect on in vitro or in vivo VEGF-A–induced angiogenesis, it prevented VEGF-A–induced retinal hyperpermeability in rats to the same extent as aflibercept (a VEGF-A neutralizing agent), a currently established treatment of DME (Sivaprasad et al., 2022). This may be due to their different mechanisms; aflibercept inhibits pathologic neovascularization in the vitreous (preretinal tufts) as a consequence of scavenging vascular growth factors like VEGF-A, whereas Nrp-1 is a denoted VEGFR2 coreceptor to which VEGF-A binds with high affinity (Appleton et al., 2007; Roth et al., 2016). Our data also show that, unlike BI-Y, administration of aflibercept does not affect tip cell density and instead numerically increased avascular area size (i.e., worsened ischemia). The significant difference between BI-Y and aflibercept illustrates the superiority of BI-Y on these parameters.
Taken together, these data demonstrate that BI-Y binds to Nrp-1 and antagonizes its biologic effects, promoting retinal revascularization and preventing VEGF-A–induced retinal hyperpermeability. This builds upon previous research by indicating that modulation of Nrp-1 may not only ameliorate diseases originating from pathologic neovascularization or hyperpermeability (Fernández-Robredo et al., 2017) but also ischemia. BI-Y may therefore have a clinical benefit in eye diseases such as DMI by promoting physiologic revascularization and preventing pathologic neovascularization, and in DME by preventing hyperpermeability without negatively affecting angiogenesis (which could exacerbate comorbid conditions such as DMI). Given that some work has suggested that the treatment of retinal diseases with anti–VEGF-As may not improve ischemia (Chung et al., 2008; Nakamura et al., 2012), an alternative treatment that may simultaneously improve both edema and ischemia is promising.
Patients with proliferative DR typically have severe macular ischemia and central vision loss, making them an appropriate population in which to investigate treatments such as BI-Y that could address the underlying causes of vision-threatening ischemia (Sim et al., 2013; Wong et al., 2018).
In summary, BI-Y specifically binds to human Nrp-1 without competing with VEGF-A165 and inhibits Sema3A-induced cytoskeletal collapse in vitro. In vivo, BI-Y may enhance revascularization of ischemic areas in an OIR mouse model and prevent VEGF-A–induced retinal hyperpermeability in rats without interfering with VEGF-A–dependent choroidal neovascularization. Taken together, the results of the preclinical studies presented here support further investigation of BI-Y as a potential treatment of DMI and DME.
AcknowledgmentsThe authors thank Christoph Reuss, Sebastian Eder, Nicole Ernst, and Petra Wieland for expert technical assistance in cellular assays. The expert technical assistance of Monika Krauth, Dennis Schaefer, and Paula Thomanek is acknowledged. Image analysis of avascular area in the OIR model was performed by Gerald Birk. Analysis of preretinal nuclei in histological sections of the OIR model was done by Tanja Schönberger and Birgit Stierstorfer.
Authorship ContributionsParticipated in research design: Thomas, Low, Hansen, Bakker, Zippel.
Conducted experiments: Thomas, Low, Hansen, Zippel.
Performed data analysis: Thomas, Low, Hansen, Zippel.
Wrote or contributed to the writing of the manuscript: Thomas, Low, Hansen, Bakker, Zippel.
FootnotesReceived October 11, 2022.Accepted February 21, 2023.Funding for this study was provided by Boehringer Ingelheim Pharma GmbH & Co. KG (Biberach, Germany). This work received no external funding. Medical writing support was provided by Imogen Allred, DPhil, of OPEN Health Communications (London, UK) and funded by Boehringer Ingelheim.
The authors have declared a conflict of interest. All authors are employees of Boehringer Ingelheim.
dx.doi.org/10.1124/jpet.122.001473.
↵ This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
留言 (0)