
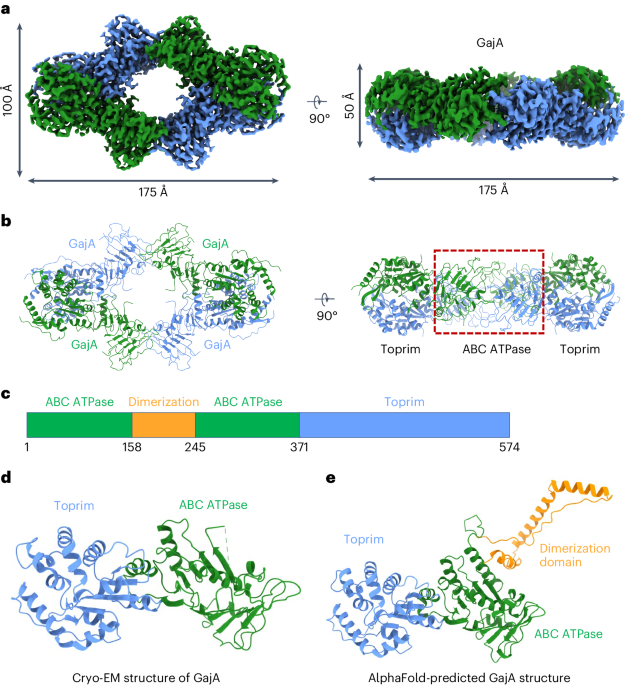
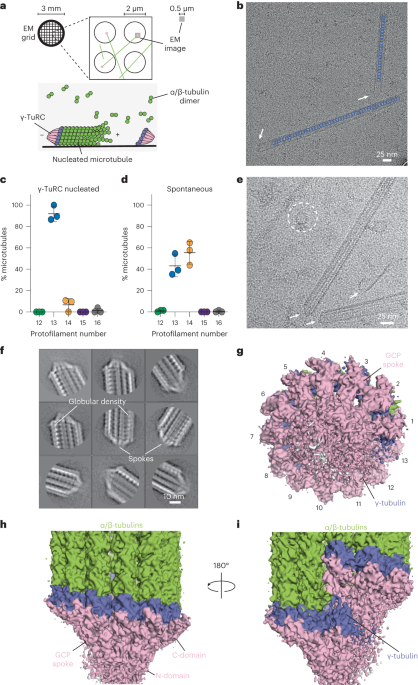
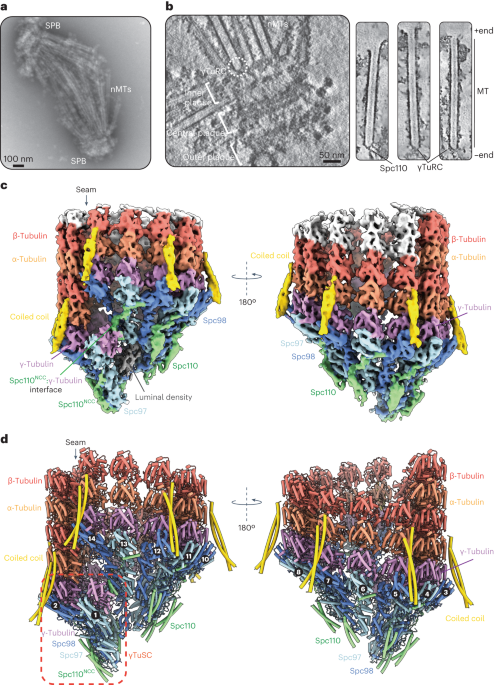
記住我

We set out to functionally assess the RNA regulatory activity of proteins across the entire yeast proteome through the tethered-function assay. Flow cytometry provides high-throughput, single-cell phenotypic measurements and enables large, pooled screens using fluorescence-activated cell sorting (FACS). FACS analysis of tethered-function assays relies on fluorescent protein reporters17, and so we devised a budding yeast tethering assay coupled to a ratiometric fluorescence readout. We tethered a transcript encoding a yellow fluorescent protein (YFP) with five boxB hairpins in its 3′ untranslated region (UTR) to a candidate regulatory protein fused to the λN coat protein18. To control for non-specific changes in cell size and physiology, we normalized the YFP measurements against a red fluorescent protein (RFP) control expressed from a transcript that is not targeted by λN. Changes in the ratio of fluorescence intensity between the yellow reporter and the red control precisely measure specific regulatory activity affecting the targeted mRNA while controlling for global effects (Fig. 1a). To further control for the possibility that binding of λN itself affects the reporter, we normalized the fluorescence ratio of the tethered fusion constructs against a tethered HaloTag protein, which exhibits no inherent regulatory effect.
Fig. 1: The dual reporter tethering assay reports reproducible and quantitative regulatory effects.
a, Schematic of the tethering assay with a YFP reporter and RFP control (top), with expected fluorescence levels based on the activity of the tethered query protein (bottom). b, Schematic of testing the effects of the reporter mRNA and protein-RNA tether. c, The fluorescence ratio changes from tethering Pab1 and Pop2 to fluorescent protein reporter mRNAs, relative to tethering an inactive Halo control. Multiple regression (adjusted R2 = 0.996, F(7, 8) = 563.2, P = 3.9 × 10−10) indicated significant effects of Pab1 (log2 change of 1.35, 95% CI 1.18–1.53, t = 17.8, P < 0.001), Pop2 (log2 change of −2.40, 95% CI −2.58 to −2.23, t = −31.7, P < 0.001) and tether choice on Pop2 (log2 change of 0.92, 95% CI 0.57–1.26, t = 6.0, P < 0.001); no other terms were significant (all P values are two-sided with no multiple testing correction). d, Distribution of fluorescence ratios reporting on the activity of Sgn1 in the tethering assay in two replicate samples. The dashed line represents the median YFP expression.
We validated our assay by measuring how well characterized regulators affected reporter expression. Tethered poly(A)-binding protein (Pab1 in budding yeast) enhances reporter expression by stabilizing mRNA14 and promoting its translation19. We observed an approximately threefold target RNA activation by tethered Pab1-λN, relative to an inactive HaloTag-λN control. Conversely, the CCR4–NOT complex is responsible for the majority of cytosolic mRNA deadenylation20, and tethering of the CAF1 deadenylase (Pop2 in budding yeast) greatly destabilizes target mRNAs21. We saw approximately fivefold reporter repression by tethered Pop2-λN (Fig. 1b,c). We further tested how the particular choice of the λN•boxB interaction pair affected our results by tethering Pab1 and Pop2 to reporters containing one PP7 hairpin using fusions with the PP7 coat protein (PP7cp)18 (Fig. 1b and Extended Data Fig. 1a). Both PP7cp fusions showed similar activity on their cognate targets as λN, although Pop2-PP7cp repression appeared weaker than Pop2-λN repression, potentially due to the use of only a single PP7 hairpin (Fig. 1c). We went on to measure the activity of the RBP Sgn1, which is linked to translation by genetic interactions and co-immunoprecipitation with Pab1 (Extended Data Fig. 1b)22. We found that Sgn1 served as a powerful activator that upregulated YFP expression by over sixfold relative to RFP (Fig. 1d), in addition to modestly increasing RFP levels and cell size (Extended Data Fig. 1c–f). Sgn1 tethering increased YFP RNA abundance ~2.5-fold (Extended Data Fig. 1g); based on the larger change we see in YFP fluorescence, we infer that it activates translation as well. These results confirm that our tethering assay provides robust and quantitative measurements of mRNA-specific regulatory activity, even in the face of additional non-specific effects on the cell, and thus provides a powerful tool for a high-throughput, proteome-wide survey of mRNA regulators.
A proteome-wide survey of post-transcriptional regulatorsWe set out to comprehensively survey the yeast proteome for post-transcriptional regulators by creating a large pool of cells that each expressed one λN fusion construct, sorting these cells into subpopulations according to their fluorescence phenotypes, and quantifying the tethering constructs in each of these sorted groups by deep sequencing. Tethering protein fusions with regulatory activity would alter the fluorescence phenotype of the host cell, shifting it into a subpopulation with an unusually low or high fluorescence ratio (Fig. 1a), and altering its distribution across the sorted cells.
We began by generating a proteome-scale library of λN fusions that would enable unbiased discovery of regulatory proteins and identification of functional domains within these regulators. We reasoned that we could construct an unbiased λN fusion library directly from randomly fragmented genomic DNA as budding yeasts have a compact and intron-poor genome, and thereby obtain a uniform representation of all proteins. However, we required an additional selection for fragments matching the correct strand and frame of a gene. We generated fragments by transposon-mediated tagmentation23,24 and selected fragments of ~500 base pairs to capture whole protein domains, which have a typical size of ~100 amino acids25 (Fig. 2a and Extended Data Fig. 2a). We captured these fragments into a vector that required in-frame translation through the fragmented sequence to express a downstream selectable marker (Fig. 2b). We found that ten out of ten individual clones encoded in-frame fusions (Supplementary Data 1). We then transferred our fragment library into a λN fusion expression vector and added random, 25-nucleotide barcodes that identify each fragment uniquely (Supplementary Data 2)26,27. The mean fragment size in our barcoded λN fusion library was ~500 base pairs, consistent with the fragment size of the genomic DNA input (Fig. 2c), and contained at least one representative fragment from roughly half of all yeast genes.
Fig. 2: Generating an unbiased, proteome-wide survey of tethered in-frame protein fragments.
a, S288C genomic DNA was fragmented by transposon-mediated tagmentation and selected to recover fragments with an average size of 500 base pairs. b, DNA fragments were cloned by in vivo gap repair into a plasmid containing a downstream selectable marker. Fragments containing an open reading frame in the correct phase will express a functional Schizosaccharomyces pombe HIS5 gene and support growth on selective media, whereas cells harboring out-of-frame fragments will fail to grow. c, Selected fragments were subcloned into the tethering vector with the λN and blue fluorescent protein (BFP) proteins encoded downstream. Fragments were assigned on average three barcodes each. The barcoded library size distribution did not change significantly from in the initial input fragment library. d, The library was transformed into the dual reporter yeast strain where the fragments were tethered to YFP mRNA. Phenotypic changes were captured by FACS sorting based on YFP versus RFP expression. Plasmids encoding the tethered fragments were isolated, and the barcodes associated with each fragment were amplified and then quantified through next-generation sequencing. e, Distribution of read counts per FACS bin for the Sbp1(28–197) activator fragment. f, As in e, for the Ebs1(708–876) repressor fragment. g, Kernel density estimate (KDE) of the library-wide activity score distribution.
We analyzed the regulatory activity of each individual protein fragment in our library by pooled transformation, flow sorting and sequencing. We separated a population of cells transformed with our λN fusion library into four subpopulations of equal size according to the YFP/RFP fluorescence ratio, isolated library plasmid DNA from sorted cells, and quantified the barcodes by next-generation sequencing (Fig. 2d). We expected activators to be enriched in bins with higher YFP/RFP ratios, while repressors should be enriched in bins with lower ratios.
Indeed, certain tethering constructs displayed a dramatic skew in their abundance across the sorted cells. For example, one fragment of the RBP Sbp1 was sorted almost entirely into the highest YFP gate, indicating that it strongly activated reporter expression (Fig. 2e). We saw a similar strong enrichment for fragments of Pab1 (Extended Data Fig. 2b), reproducing the positive effect of tethering full-length Pab1 (Fig. 1c). Conversely, fragments of the nonsense-mediated decay factor Ebs1 and the RNA destabilizing protein Cth1 acted as strong repressors that were found almost exclusively in the lowest YFP subpopulation (Fig. 2f and Extended Data Fig. 2c). To quantify this enrichment, we computed an ‘activity score’ for each fragment: a maximum likelihood estimate of its average fluorescence, expressed as a z-score relative to the overall population. These scores ranged from −1.9 for strong repressors like Ebs1 and Cth1 to +1.9 for strong activators like Sbp1 and Pab1. Most fragments in our library had activity scores close to zero, indicating little or no effect on reporter transcript expression (Fig. 2g and Supplementary Data 3). Activity scores were reproducible between two biological replicate screens (Extended Data Fig. 2d); fragments with adequate sequencing coverage (at least 1,000 total reads across all bins) in both experiments had an activity score correlation of r ≈ 0.7. We did note a linear rescaling of scores between the two screens, leading to saturation of strong activators and repressors in one replicate screen relative to the other. Because of this saturation effect, the strong correlation nonetheless underrepresents the actual agreement between the two screens. We relied on activity scores derived from the screen with broader dynamic range for our subsequent analysis.
We identified active fragments from many well-known regulatory proteins, such as the translation initiation factor Ded128,29,30 and Ngr1, which induces the decay of POR1 mRNA31. Our unbiased approach also uncovered post-transcriptional regulation in proteins with other well characterized cellular functions, including the small heat shock chaperone Hsp26, which also has previously identified mRNA-binding activity32. Furthermore, we uncovered regulatory regions in proteins of unknown function, like Her1, which may interact with ribosomes based on co-purification experiments33. These results illustrate the power of our approach to discover proteins that control mRNA stability and translational efficiency and quantify how this affects gene expression.
Full-length protein activity resembles truncated fragmentsWe selected 12 fragments across a range of activity scores and biological functions (Fig. 3a) and directly measured their effect on reporter fluorescence. All 12 fragments shifted the fluorescence ratio in the direction expected from the large-scale survey (Fig. 3b), and the magnitude of the change correlated very well with their activity score (r = 0.91) (Fig. 3c and Extended Data Fig. 3a–e). This strong quantitative agreement demonstrates that the activity score derived from sorting and sequencing is an accurate measure of the regulatory effect of a fragment.
Fig. 3: Protein fragment activity in the tethering screen represents real, verifiable regulatory function.
a, Distribution of sequencing reads across subpopulations separated by FACS. b, Median activity of each protein fragment in the flow cytometry tethering assay (n = 2 per fragment). c, Comparison of the log2(difference in fluorescence ratio) and the screen activity score per fragment, r = 0.91. d, Flow cytometry measuring activity of Sbp1 and Sbp1(14–178) in the tethering assay (n = 2, one replicate per sample is shown). e, As in d, for Sro9 and Sro9(14–151) (n = 2, one replicate per sample is shown). f, As in d, for Jsn1 and Jsn1(144–295) (n = 2, one replicate per sample is shown). g, As in d, for Yap1801 and Yap1801(374–527) (n = 2, one replicate per sample is shown).
Isolated protein fragments may have different activities than the full protein from which they are derived due to the absence of regulatory domains, altered protein-protein interactions, or other reasons. We thus selected a handful of active fragments to explore how fragment activity relates to the full protein. Sbp1 is an RBP with two RNA recognition motifs (RRMs) in addition to an arginine––glycine–glycine (RGG) motif that recruits Pab134. The fragment that we characterized as an approximately threefold activator (Fig. 2e) contained only the first RRM and the RGG motif, whereas the full-length version of the protein was a weaker, approximately twofold activator (Fig. 3d). We hypothesize that the inclusion of the second RRM interferes with Pab1 recruitment, making it a weaker activator. In other cases, such as Sro9, the full-length protein had a stronger effect than the isolated fragment. Sro9 is an RBP that contains a La-motif and is hypothesized to activate translation through recruitment of the closed-loop-forming translation initiation complex35. We identified an Sro9 fragment that activated expression approximately twofold, whereas the full-length protein increased reporter expression by nearly fourfold (Fig. 3e). Tethering the entire yeast Puf-domain protein Jsn1 likewise produced a stronger repressive effect than the fragment we identified in our tethering library (Fig. 3f). In contrast, the intact version of the endocytic protein Yap180136 was less repressive than our fragment (Fig. 3g), perhaps because of differences in localization37. Nonetheless, in all four cases, the full-length protein exerted an effect in the same direction as the fragment tested in our screen. Our approach is thus well suited to survey the regulatory activity contained in the native proteome and ascribe functions to RBPs.
Activity in RBPs but not RBDsOur tethering assay can detect regulatory activity in truncated proteins lacking RBDs and in co-regulator proteins that lack intrinsic RNA-binding activity. Nonetheless, we did expect a substantial overlap between the post-transcriptional regulators detected in the screen and known RBPs. To test this hypothesis, we compiled a list of budding yeast RBPs from proteins appearing in at least two of four RNA-protein interaction datasets (Fig. 4a)9,38,39,40,41. Fragments from these known RBPs had substantially higher absolute activity scores than the overall proteome (Fig. 4b), further confirming the relevance of our results for endogenous programs of post-transcriptional regulation controlled by these RBPs. It also raised the question of whether regulatory activity was associated with the RBDs of these RBPs.
Fig. 4: Global analyses reveal enrichment of protein domains, motifs and protein-protein interactions amongst most active screen fragments.
a, Venn diagram of overlapping datasets identifying RBPs. b, KDE of absolute activity scores for RBPs, all proteins, and non-RBPs in the screen. c, Pfam protein domains significantly enriched amongst active screen fragments (adjusted P values from a two-sided Mann–Whitney U test with Benjamini–Hochberg multiple testing correction). d, Plot of proteins significantly enriched in interactions with activator fragments, where the x axis represents fold enrichment. e, Pab1 interactions with activator fragments (outer ring), with overlapping Gis2 interactions depicted in red.
Our fragment library allowed us to ascribe quantitative regulatory effects to particular regions and domains within proteins. We were thus able to investigate which protein domains were enriched among the most active fragments in our screen, and whether these active regions coincided with RBDs. We identified fragments that contained at least 75% of some protein domain family from the Pfam database42 and tested each family individually to determine whether the activity scores of fragments containing that family were significantly higher or lower than the library overall (Fig. 4c).
Dozens of protein families were associated with active regulators, and some of the strongest associations involved domains with clear connections to translation and RNA decay (Supplementary Data 6). We observed the strongest positive mean activity score among fragments derived from the translation initiation factor eIF343. We also saw a trend for activators among that DEAD box helicase family proteins, which include the translation initiation factors eIF4A and Ded144. The endo/exonuclease/phosphatase family showed up among the strong repressors; these include certain subunits of the Ccr4–Not complex, for example42. We also saw many families encoding metabolic functions such as adenylosuccinate synthetase45, FAD-dependent oxidoreductase and the malic enzyme N-terminal domain. Metabolic enzymes have emerged as cryptic RBPs9, and so it seems noteworthy that they appear to show regulatory activity as well. Notably, although many canonical RBDs such as RRMs appear in Pfam, they were not enriched in the active fragments. Canonical RNA-recognition domains appear more important for mRNA target selection, and regions outside of the RNA-interacting domains typically provide regulatory activity for RBPs.
Our screen also identified strong activity in fragments lacking an identifiable, folded domain. Indeed, many proteins contain intrinsically disordered regions (IDRs), which play important roles in post-transcriptional regulation46. In some cases, IDRs form protein-protein interactions, as in the case of the disordered N terminus of Ded147,48, whereas others serve as flexible linkers49,50. Functional IDRs can include short linear interaction motifs (SLiMs), which are often responsible for protein-protein interactions51. Although SLiMs are distinct from Pfam domains, they may be recognizable as peptide sequence motifs.
Motivated by the possibility that SLiMs could explain regulatory effects, we searched for peptide motifs enriched in active fragments using the MEME tool, and then scanned the yeast proteome for occurrences of these motifs using FIMO52. Some motifs were highly repetitive; although these repetitive motifs may have regulatory activity, it is difficult to interpret them, so they were excluded. We identified six non-degenerate motifs and repressors (Extended Data Fig. 4a,b and Supplementary Data 7 and 8), which align to genes with functions spanning many aspects of cell biology, including cell wall maintenance, cytoskeleton functions, transcription and translation. The glutamine-rich motif (repressor motif 2 in Extended Data Fig. 4a) is particularly enriched in genes involved in mRNA metabolism, such as NGR1, POP2 and PUF3, which all have diverse roles in mRNA deadenylation and decay31,53,54. Likewise, the RGG repeat in activator motif 5 (Extended Data Fig. 4b) is widespread among RBPs and is linked to post-transcriptional regulation55.
Regulatory RBPs often exert their effects by recruiting and activating core cellular machinery involved in translation and RNA decay. We thus expected that distinct active fragments from our screen might share common interactors. We intersected our library fragments with the physical protein-protein interactions in the BioGRID database56 and searched for proteins with a significant over-representation of activating or repressing fragments among their interactors. We identified a dozen proteins enriched for interaction with activators (Supplementary Data 9), most tied clearly to RNA biology (Fig. 4d). Strikingly, the poly-(A) binding protein Pab1 showed one of the highest degrees of enrichment19,57. The translation regulator Gis258,59 was also substantially enriched in activators, and shared many interaction partners with Pab1 (Fig. 4e). Surprisingly, the exonuclease Xrn1 exhibited the strongest enrichment in activator interactions (Fig. 4d), despite its role in mRNA decay60. This enrichment may reflect a common core of mRNA-binding proteins that accompany transcripts during both translation and degradation. Alternatively, Xrn1 is reported to promote the translation of some transcripts encoding membrane proteins, and so this enrichment might also represent a more direct effect61.
Endoplasmic reticulum/Golgi protein Gta1 is a bimodal repressorSeveral overlapping, C-terminal fragments of the protein Gta1 harboring a repressor-associated peptide emerged as potent repressors (Figs. 4b and 5a and Supplementary Data 5). Although the Gta1 protein co-purifies with the translational machinery33, genetic evidence links it to golgi and vesicle transport62,63, and the Gta1-GFP fusion protein localizes to the endoplasmic reticulum (ER)64,65,66. Owing to its reported association with ribosomes and the presence of a repressive motif, we generated λN fusion constructs of the strongly repressive Gta1(603–767) fragment and the full-length Gta1 protein, and tested their effects on reporter expression (Fig. 5b).
Fig. 5: The tethering screen identifies RNA-regulatory roles of poorly characterized proteins.
a, Schematic representation of the Gta1 protein, with the C-terminal Gta1(603–767) fragment highlighted. b, Schematic depiction of Gta1(603–767) in the tethering assay. c, Flow cytometry measuring activity of Gta1 and Gta1(603–767) in the tethering assay, where dashed lines represent the median YFP expression (n = 2, one replicate per sample is shown). d, RT–qPCR analysis of YFP mRNA abundance with Gta1 tethered to the 3′ UTR, normalized to a non-regulator control (n = 3 independent biological replicates are shown in different shades; P = 0.00026, two-sample t-test with unequal variance). e–g, Time course of reporter changes after induction of Gta1 (e), Gta1Δ603–767 (f) and Halo control tethering constructs (n = 2) (g). h, Change in BFP fluorescence as a measure of Gta1 expression over time, normalized to the uninduced Gta1 sample (n = 2, one replicate is shown per time point). i, As in h, for Gta1Δ603–767. j, RT–qPCR analysis of induced GTA1 relative to endogenous GTA1 expression (n = 3 biological replicate cultures). k, Light microscopy images of yeast overexpressing GTA1, GTA1Δ603–767 or Halo control for 4 h (n = 3 biological replicate cultures are shown, with two frames per replicate). Scale bar, 20 µm.
Both full-length Gta1 and the Gta1(603–767) fragment robustly reduced median YFP and produced a strongly bimodal distribution of reporter expression (Fig. 5c and Extended Data Fig. 5a), a distinctive pattern that we did not see for any other tethering construct we examined individually. As expression of the isolated Gta1(603–767) fragment slowed cell growth, we focused our analysis on full-length Gta1. Gta1 tethering greatly reduced reporter mRNA abundance (Fig. 5d), suggesting that it promoted mRNA turnover. To track how bimodality emerged when the Gta1-λN fusion was switched on acutely, we expressed it from an inducible promoter. Levels of the YFP reporter began to decline within 1 h of inducing the tethering construct, and clear bimodality emerged within 2 h (Fig. 5e); continuing decline of reporter levels in the lower peak probably reflects the loss of pre-existing YFP through degradation or dilution. Notably, deletion of the repressor fragment that we identified in a Gta1Δ603–767-λN tethering construct abolishes this effect entirely (Fig. 5f,g), confirming that the Gta1(603–767) region containing our repressive peptide motif is both necessary and sufficient for its regulatory effect.
We next tested whether the bimodal reporter expression resulted from variation in the abundance of the Gta1 tethering fusion. Indeed, we saw a broad, bimodal distribution of blue fluorescent protein (BFP) fluorescence from the Gta1-BFP-λN construct after 4 h of induction (Fig. 5h), with levels increasing uniformly in the first hour of induction, followed by the emergence of two distinct phenotypes (Fig. 5h). Notably, we saw a similar trajectory after induction of the Gta1Δ603–767-λN tethering construct (Fig.
留言 (0)