
記住我

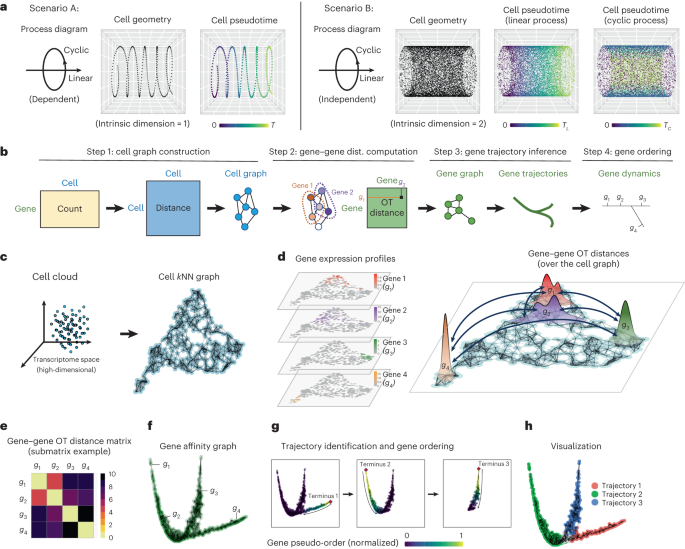
The input to StabMap is a set of s appropriately scaled and normalized data matrices, \(}}\}_,}_,\ldots ,}_}\}\), not necessarily containing the same features, and optional discrete cell labels for any of the datasets. As an initial step, StabMap generates the corresponding MDT. The MDT is an undirected weighted network that contains s nodes, one corresponding to each data matrix, with edges being drawn between pairs of nodes for which the corresponding data matrices share at least one feature. The edges in the MDT are weighted according to the absolute number of common features between the two datasets. StabMap requires that the MDT be a connected network, that is, that there exists a path between any two nodes. Weighted shortest paths are calculated between any two given nodes in the MDT.
The StabMap algorithmAt least one dataset must be considered as a reference dataset, with the option for multiple datasets to be considered as reference datasets. The output of StabMap is a common low-dimensional embedding with rows corresponding to all cells across all datasets, and columns corresponding to the sum of lower dimensions across the reference dataset(s). For a reference dataset Dr, two matrices are extracted, first a scores matrix Sr (a cells × low-dimensions matrix) and a loadings matrix Ar (a features × low-dimensions matrix) such that \(_}=_}^\times _}\). If no cell labels are provided, principal components analysis (default 50 PCs) is used for estimation of Sr (as the PC scores) and Ar (the components loadings). Alternatively, if discrete cell labels are provided, linear discriminant analysis is used for estimation of Sr (as the linear discriminants for each class) and Ar (the feature discriminant loadings).
Then, for each of the s data matrices, score matrices \(}_^},}_^},\ldots ,}_}^}\) are calculated in one of the following ways for data matrix i:
If i = r, then the scores matrix Sr is returned, that is, \(}_}^}=}_}\).
If i and r share an edge in the MDT, and all features in Ar are present in Di, then \(}_}^}\) is directly calculated as the projected scores, that is \(_^=_^\times _\), where Xi is the appropriate submatrix of Di to match the features in Ar. If not all of the features in Ar are present in Di, then \(_^\) is estimated using multivariate linear regression on each column of Sr for dataset Dr. Specifically, for column j of Sr, we fit the model \(_\left[\,j\right]=_\left[\,j\right]_\left[\,j\right]+}\) where \(}_, > }\) is the submatrix of Dr for features that are shared among Di and Dr, and ϵ is assumed to be normally distributed noise. \(_\) therefore is a matrix of fitted coefficients \(\left(_, > ,1}},\ldots ,_, > ,}},\ldots \right)\) with rows corresponding to the shared features between Di and Dr and columns corresponding to the columns of Sr. The estimated score matrix for i is taken to be the predicted values of the multivariable linear model for dataset Di, and is calculated as \(}_}^}=}_, > }}_, > }\) where \(}_, > }\) is the submatrix of Di for features that are shared among Di and Dr.
If i and r do not share an edge in the MDT, then \(_^\) is estimated using an iterative approach that exploits the shortest weighted path in the MDT. Starting from node r, for the next node along the path p, we calculate \(}_}^}\) as described above. If the next node along the path is i, then we fit the model \(_^\left[j\right]=_\left[j\right]_\left[j\right]+\epsilon\) where \(}_, > }\) is the submatrix of Dp for features that are shared among Dp and Di and \(}_, > }\) is the matrix of fitted coefficients \(\left(}_,\ldots ,}_,\ldots \right)\). The estimated score matrix for i is then taken as the predicted values of this multivariable linear model for dataset Di, and is calculated as \(}_}^}=}_, > }}_, > }\). If instead, the next node along the path from r to p and eventually to i is some other node q, then this process of fitting a multivariable linear model and predicting on the new data is repeated until we calculate \(}_}^}=}_, > }}_, > }\), where w is the node previous to q along the path between r and i.
The estimated score matrices for each of the s datasets are then concatenated across rows to form the joint low-dimensional score where reference r is employed: \(}^}=\left(}_^},}_^},\ldots ,}_}^}\right)\), where Sr is a matrix with number of rows equal to the total number of cells across all s datasets and number of columns equal to the number of columns (selected features) in Sr.
We believe StabMap’s improved performance over naive approaches can be explained by noting that the features that drive biological variation may either not be captured, or represent the dominant signal, in the shared feature space, and are therefore not prioritized when reducing dimensionality using PCA on the shared features. StabMap’s linear regression strategy estimates the linear combination of the shared features that best captures the (assumed to be) biological variation that is dominant in the full feature data.
StabMap with multiple reference datasetsFor the set of reference datasets R = ⊆ D, we calculate the corresponding set of joint low-dimensional scores as described above, S = . We reweight each scores matrix S j according to the overall L1 norm of the matrix and a user-set weighting parameter \(}_}\in \left[0,1\right]\) (by default set to 1),
$$}^* }=}_}\frac}^}}_}\left|}^}\right|}.$$
The user-set weighting parameter wj controls the magnitude of the score vectors for each reference dataset, and thus corresponds to the relative influence of the reference dataset on any magnitude-based downstream analysis (for example, calculation of Euclidean distances between cells). To generate common low-dimensional scores across all reference datasets, we concatenate the reweighted scores across columns to form the StabMap low-dimensional scores, \(=\left(}^}_}}}^}_}}\ldots \right)\) for reference data indices j1, j2,…. S is a matrix with number of rows equal to the total number of cells across all s datasets, and number of columns equal to the total number of columns across the scores matrix for each reference dataset.
StabMap computational speedStabMap takes on the order of seconds to less than a minute for tens of thousands of cells on a standard MacBook. We observed StabMap taking on the order of 5–10 min running for 300,000 cells in our breast cancer analysis. We believe this speed can be attributed to several aspects of the software implementation. PCA is performed via the fast irlba algorithm, linear model fits are performed using the underlying R machinery via lm.fit, therefore reducing time and memory costs, and finally we retain the use of sparse matrix representation of data at every opportunity we can. While we use R’s native vectorization to speed up computation, one memory limitation at present is the need to convert to dense matrix representations for imputeEmbedding, this is due to the dependency of ‘abind’ package in R that works only for dense matrices. Future work could incorporate some sparse 3D array representation, thereby circumventing the need to convert data into dense matrices, or potentially to harness the capability of delayed matrix operations without needing to load data into memory. We find that runtime increases with the number of input datasets, as well as the proportion of datasets to be considered as references, as mapping across the MDT is repeated for each selected reference dataset.
Downstream analysis with StabMapBatch correctionWhile StabMap jointly embeds cells across multiple datasets into a common low-dimensional space, batch effects both within and among datasets can remain. Any existing batch correction algorithm that works on a low-dimensional matrix (for example, fastMNN20, scMerge22 and BBKNN39) can be employed to obtain batch-corrected StabMap embeddings. In the analyses presented in this manuscript we use fastMNN as downstream horizontal data integration. For the simulation presented in Fig. 2, we perform two additional horizontal data integrations using Harmony26 and Seurat21. For the latter case we treat the StabMap low-dimensional features as input features to Seurat, with parameters adjusted to not perform any feature selection or further dimensionality reduction.
Supervised and unsupervised learningThe batch-corrected StabMap embedding facilitates supervised learning tasks such as classification of discrete cell labels using any suitable method such as k-nearest neighbors, random forest and support vector machines, and regression using traditional linear models or support vector regression. Unsupervised learning tasks can be performed by clustering directly on the embedding (for example, k-means clustering) or by first estimating a cell–cell graph (for example, shared nearest neighbor or k-nearest neighbor graph) followed by graph-based clustering (for example, Louvain or Leiden graph clustering). Since one can use the embedding to estimate the cell–cell graph, additional bespoke single-cell analyses such as local differential abundance testing between experimental groups, such as that implemented in Milo40, can be employed.
Imputation of original featuresWe include an imputation implementation based on the StabMap low-dimensional embeddings to predict the full-feature matrices for all data, by extracting the set of k neighbors using Euclidean distance within the StabMap-projected space and returning the mean among the nearest neighbors. This is especially useful for projecting query data onto a reference space or for identifying informative features downstream of the data integration step.
Mosaic data integration simulationsWe used publicly available data to investigate the performance of StabMap and other methods, as described below.
PBMC 10x Multiome dataWe used the SingleCellMultiModal R/Bioconductor package41 to download the ‘pbmc_10x’ dataset, containing gene expression counts matrix and read counts associated with chromatin peaks captured in the same set of cells. We normalized the gene expression values using logNormCounts42 in the scuttle package, and restricted further analysis to HVGs selected using the ModelGeneVar function in scran43. For the chromatin data modality we performed term frequency—inverse document frequency (TF-IDF) normalization according to the method described in ref. 10. We extracted peak annotation information using the MOFA2 R package tutorial18, including information on which genes’ promoters the chromatin peaks were associated with, if any. These promoter peaks were annotated as the associated gene name, so that the promoter peak features would match the RNA genes features.
To perform the mosaic data integration simulation with the PBMC 10x Multiome data, we ignored the matched structure between the RNA and chromatin modalities, and treated this data as if they belonged to two distinct datasets. We performed StabMap using both RNA and chromatin modalities as the reference datasets, and reweighted the embedding to give equal contribution for the two modalities. For assessing the cell type accuracy we used the RNA modality cells as labeled data, and predicted the cell types of the chromatin modality cells using k-nearest neighbors classification with k = 5.
Mouse Gastrulation Atlas scRNA-seqWe downloaded the counts data from Pijuan Sala et al. (2019)1 using the MouseGastrulationData R/Bioconductor package44 corresponding to E8.5, and normalized in the same way as the 10x Multiome PBMC data. Then, we split the dataset into four groups according to the four sequencing samples. For each randomly selected pair of sequencing samples, we artificially assigned one sequencing sample as the query dataset and kept one other sequencing sample intact as the reference dataset. Within each simulation round, we performed HVG selection from the reference dataset, and randomly selected 50, 100, 250, 500, 1,000, 2,000 and 5,000 genes to be kept for the query dataset.
We used StabMap to jointly embed the reference and query datasets into a common low-dimensional space by selecting the reference dataset as the sole reference, followed by batch correction using fastMNN. We also performed naive PCA, UINMF and MultiMAP for comparison. To assess performance, we calculated the mean accuracy of cell type classification of query cells using k-nearest neighbors with k = 5 for each method.
To assess the effect of downstream horizontal integration on embeddings using StabMap and naive PCA, we performed additional batch correction algorithms Harmony, fastMNN, and Seurat on the embeddings, as well as retaining uncorrected embeddings. We then calculated the difference in cell type accuracy between StabMap and naive PCA for each of the simulation scenarios and batch correction algorithms.
PBMC CyTOF dataWe downloaded the PBMC CyTOF27 data using the HDCytoData45 package in Bioconductor. This dataset included two conditions of stimulated and unstimulated PBMCs from healthy individuals, of which we selected only unstimulated control cells for further analysis. From this data we extracted 24 protein features corresponding to biologically relevant signal.
PBMC ECCITE-seq dataWe downloaded the PBMC ECCITE-seq data28 using the SingleCellMultiModal41 package in Bioconductor. This dataset included control and treated samples, from which we selected only control samples for further analysis. For these data, we extracted the single-cell RNA component and the cell surface ADT protein data.
Breast cancer IMC dataWe downloaded the processed breast cancer IMC data29 using the Zenodo link provided in the publication. We selected only samples that corresponded to patients with positive estrogen receptor (ER) status and PAM50 classification of HER2, resulting in a set of 32,400 IMC-resolved cells, for which 37 protein features were profiled.
Breast cancer CITE-seq dataWe downloaded the processed breast cancer CITE-seq data30 via GEO and the Broad Institute single-cell portal links provided in the publication. We selected a single patient sample, corresponding to an HER2-positive case. Then we combined the RNA and ADT modalities into a single data object using CiteFuse preprocessing tool46.
Breast cancer spatial transcriptomic dataWe downloaded the processed breast cancer Xenium data31 on 3 November 2022 from the 10x Genomics website provided in the publication. We retained cells that captured at least 30 transcripts, and performed standardization using logNormCounts, resulting in a genes by cell expression matrix.
Comparison with other methods UINMFWe used software version 0.5.0 of LIGER, which includes the UINMF implementation, and performed integration using defaults as suggested in the LIGER vignette. We used the counts matrix for input, as suggested in the vignette. We used the resulting 50-dimensional embedding for subsequent downstream analysis, and uniform manifold approximation and projection (UMAP) implemented in scater42 for visualization.
MultiMAPWe used the Python (version 3.8.10) package MultiMAP (version 0.0.1), and performed data integration using defaults as suggested by the MultiMAP tutorial website with equal weights for each dataset. The output of MultiMAP is a corrected graph representation, as well as a two-dimensional representation of the data. We used this two-dimensional representation for visualization and to perform downstream analysis tasks.
Naive PCATo implement naive PCA, we first extracted the submatrices of datasets containing features that were common across all datasets. We then performed PCA using scran’s implementation with 50 principal components, followed by batch correction using MNN. We used the 50-dimensional representation for downstream analysis tasks, and UMAP to perform further dimensionality reduction to two dimensions for visualization.
CoboltWe used the Python (version 3.8.10) package Cobolt (version 0.0.1), and performed data integration using defaults as suggested by the tutorial, with input data corresponding to the original counts for scRNA-seq gene expression and for ATAC detected open chromatin fragments. The output of Cobolt is a low-dimensional representation, which we further summarized using UMAP for visualization.
MultiVIWe used the Python (version 3.8.10) package scvi (version 0.16.4) and performed data integration using defaults as suggested by the package tutorial, with input datasets corresponding to original counts from scRNA-seq and ATAC seq multiomics, scRNA-seq and scATAC-seq. We extracted MultiVI latent space representation values, and performed UMAP for further visualization.
EvaluationTo evaluate the mosaic data integration simulations, we used three quantitative metrics.
Cell type classification accuracyGiven a joint embedding, we perform a simulation such that discrete class labels corresponding to cell types are artificially removed for a subset of the data. We then perform k-nearest neighbors classification (k = 5) to obtain the predicted class label for the artificially unlabeled data. The cell type classification accuracy is thus the proportion of cells for which the classification is correct compared to the true cell type label,
\(A=\frac_I\_^}}}=_^}}}\}}_1}\).
Jaccard similarityFor cell i in embedding S we have l positions for the l omics levels (for example, RNA and chromatin). We extract the sets of size k (default 100) containing the nearest cells of the same omics layer, that is, Nil = }_}=\text\left(}_1},}_2}\right)=\frac}_1}\cap }_2}\right|}}_1}\cup }_2}\right|}\).
Larger values of Ji correspond to larger overlap of neighbors between the two omics layers and are thus desired.
Number of nearest cells metricSimilar to the metric employed by Kriebel et al. and Jain et al.11,12, for cell i belonging to omics layer 1 (for example, RNA) in embedding S, we calculate the number of cells among omics layer 2 (for example, chromatin) that are nearer than cell i belonging to omics layer 2, \(}_2}=_}\\left(}_1},}_2}\right)\le \left(}_1},}_2}\right)\}\).
We then extract the empirical cumulative distribution of nearest cells by calculating, for each integer x, the number of cells for which their number of nearest cells metric is at most this value, \(\left(\right)=_}\}_2}\le \}\). Higher values of M(x) across all values of x are more desired.
Multi-hop mosaic data integration simulationWe used the PBMC 10x Multiome data to evaluate StabMap under the situation of multi-hop mosaic data integration. We downloaded and processed the data as described in the subsection above, with the exception that promoter peaks corresponding to specific genes were not matched to the associated genes. This resulted in a complete lack of overlap between features between the RNA and chromatin modalities.
To perform the simulation, we randomly allocated each cell into one of three classes: (1) RNA only, (2) chromatin only and (3) Multiome, with varying relative proportions of cells associated with the Multiome class. Cells within the RNA class had their chromatin information ignored, and cells within the chromatin class had their RNA information ignored, while cells within the Multiome class were left unchanged. We then used StabMap to integrate these three simulated datasets and generate a low-dimensional embedding for each simulation setting. Comparison with other methods is not possible since PCA, UINMF and MultiMAP require at least some overlapping features across all datasets.
To evaluate the multi-hop mosaic data integration simulation, we calculated the LISI26 using both modality and cell type as the grouping variables. Higher LISI values correspond to more local mixing of cells, and so relatively high values for modality and low values for cell type are desirable.
Multi-hop mosaic data integration of CyTOF, ECCITE-seq and 10x Multiome dataWe used three data sources to examine StabMap’s capability of performing multi-hop mosaic data integration. We performed matching of protein IDs between the CyTOF and ECCITE-seq datasets, resulting in an overlap of seven proteins captured by each technology. For each dataset, we reassigned cell type labels to broad common cell types including B, CD4 T, CD8 T, dendritic cell (DC), MAIT T, monocyte, natural killer (NK) and surface cells. Then, we performed StabMap using three configurations. First, using the CyTOF dataset as the reference, with the underlying number of principal components set to 10 due to the limited number of proteins captured; second, using the 10x Multiome data as the reference; and third using both as references with equal weighting. In each case we performed downstream horizontal data integration using FastMNN. We visualized the resulting StabMap embeddings using UMAP. To assess the quality of each embedding, we used the LISI metric and examined the distribution of such values among the CyTOF and Multiome cells.
Multi-hop mosaic data integration of IMC, CITE-seq and 10x Genomics Xenium dataWe used three data sources to examine StabMap’s ability to perform data integration, especially over multiple spatial omics technologies. We performed matching of protein ADT IDs between the IMC and CITE-seq datasets, resulting in 19 shared features. For the IMC and CITE-seq datasets, we reassigned cell type labels to broad common cell types including B cells, endothelial cells, epithelial cells, fibroblasts, myeloid cells, NK cells, plasmablasts, Panton-Valentine Leukocidin (PVL), T cells and tumor microenvironment (TME) cycling cells. Then we performed StabMap, selecting IMC and Xenium datasets as references, with 10 and 50 principal components respectively. Given the joint embedding extracted using StabMap, we then predicted epithelial cell class on the Xenium data, using the IMC-resolved cells as training data. Additionally, we performed feature imputation on the Xenium data, using the IMC-resolved data as training, using the imputeEmbedding function in the StabMap software. Finally, we predicted broad cell types on the Xenium data using the IMC-resolved cells as training data, and generated cell–cell contact maps (as previously described7) on two selected regions, corresponding to triple-positive receptor region, and an invasive region.
Simulation of multi-hop mosaic data integration using Mouse Gastrulation DataTo examine the capability of StabMap, we randomly selected cells from the Mouse Gastrulation Dataset described above, and split them into eight distinct datasets with varying numbers of total cells per dataset, n = 500, 1,000 and 2,000. Then, we retained varying numbers of features, n = 100, 200, 500 and 1,000 from among the HVGs such that there was approximately 50% overlap of features between datasets 1 and 2, 2 and 3, and so on. As a result, any one dataset only shared features with its neighboring dataset, representing an extreme task for multi-hop mosaic data integration. For the simulated datasets, we performed StabMap with dataset 1 selected as the reference dataset. To assess quality, we performed cell type classification (K-nearest neighbors (KNN) with k = 5) using dataset 1 as the training data and dataset 8 as the testing data, reporting the overall cell type classification accuracy as a measure of integration quality. We repeated the above simulation five times to obtain an overall mean accuracy with varying levels of number of cells and number of shared features.
Spatial mapping of mouse chimera data using StabMap scRNA-seq dataWe used the MouseGastrulationData R/Bioconductor package (Griffiths and Lun 2020)44 to download gene expression counts for the Mouse Gastrulation Atlas dataset, WT/WT control chimera dataset1, and T−/−/WT chimera dataset32, corresponding to E8.5. We combined the gene expression counts into a single dataset, then normalized and extracted HVGs using the same approach applied to the 10x Multiome PBMC data.
seqFISH dataWe downloaded seqFISH-resolved gene expression log counts7 for spatially resolved cells of mouse embryos profiled at a similar developmental stage along with their corresponding spatial coordinates. We extracted novel features for each gene g and each cell i by calculating the mean expression value among the nearest cells in space, \(}_}^=\frac_\in }_}}}_}}}_}\right|}\), where \(\)i = is the set of cells that are at most two steps away from cell i in the spatial nearest neighbor network7. We then concatenated these novel features with the measured gene expression, before downstream integration with the dissociated scRNA-seq data.
Mosaic data integration and local enrichment testingWe used StabMap, parametrized with multiple reference datasets, to integrate the scRNA-seq and seqFISH data. We used PCA (default 50 PCs) to generate the low-dimensional scores for the scRNA-seq and seqFISH references, and reweighted each scores matrix using the default weighting parameter of 1. As a result, we obtained a 100-dimensional StabMap low-dimensional scores matrix. We then corrected for any remaining batch differences using fastMNN, where batches reflect technical groups from each dataset.
To calculate whether T−/− cells were enriched in a neighborhood around each seqFISH cell, we performed logistic regression. Specifically, for each spatially resolved (seqFISH) cell, in the joint embedding we extracted its 1,000 nearest neighbors from each chimera dataset (4 T−/−/WT samples and 3 WT/WT samples), and fit the model \(}\frac}}=}}_+}}_}_+}}_}_\).
In this model, p is the vector of observed proportions of td-tomato+ cells for each chimera, x1 is a vector containing the total proportion of td-tomato+ cells belonging to a biological replicate, and x2 is a vector indicating whether a chimera is T−/−/WT or WT/WT. We extracted the estimated coefficient of interest, \(\hat}}_}\), and associated P value for each spatially resolved cell using a likelihood ratio test, resulting in a local measure of enrichment or depletion of T−/− cells for each seqFISH-profiled cell. We then used the method of Benjamini–Hochberg to calculate FDR-adjusted P values.
Mixed T−/− enrichment in pharyngeal/splanchnic mesodermTo examine the relationship between the estimated T−/− enrichment coefficient and AP axis position in the splanchnic mesoderm, we fitted principal curve models, with four degrees of freedom, for each individual spatially resolved embryo with the spatial coordinates as the underlying data33. We used the principal curve fitted values to extract the AP ranking of cells along this axis, and then used this ranking to estimate a locally smoothed T−/− enrichment coefficient along the AP axis.
To assess gene expression changes along the AP axis as T−/− cells move from being enriched to being depleted, we selected an equal number of cells anterior and posterior to the position where the smoothed T−/− enrichment coefficient is zero, and performed differential gene expression analysis using imputed gene expression values. Imputed gene expression was quantified for each spatially resolved cell using the mean gene expression value of the nearest five Mouse Gastrulation Atlas cells in the StabMap low-dimensional space. Gene expression changes along the AP axis were assessed using a nonparametric cubic splines model with three degrees of freedom along with grouping variables for the individual embryos. Statistical significance was estimated using an F-test, with a null model of no splines effects, with empirical Bayes shrinkage using the limma framework, followed by adjustment for multiple testing. For statistically significant genes, we visualized gene expression along the AP axis using local loess smoothing and ribbon plotting for the local standard error.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)