Materials
Alum Hydroxide Gel Adjuvant (10 mg/mL) was purchased from InvivoGen, and squalene was purchased from Sigma. HA and NP of H1N1 influenza virus were purchased from Sino Biological. Recombinant RBD-sc-dimers of spike protein were obtained from the Institute of Microbiology, Chinese Academy of Sciences. The NPs were purchased from VACURE and Sino Biological. Roswell Park Memorial Institute (RPMI) Medium 1640 basic, Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and phosphate-buffered saline (PBS; pH = 7.4) were purchased from GIBCO BRL (Gaithersburg, MD, USA.). Fluorescent hydrophilic dyes Cy3, Cy5, and Cy7 were purchased from Fanbo Biochemical Co., Ltd. (Beijing, China). CCK-8 was purchased from Dojindo (Kumamoto, Japan).
HEK293T cells, Huh7 cells, and Vero cells were obtained from the American Type Culture Collection (ATCC). SARS-CoV-2 pseudovirus was obtained from National Institutes for Food and Drug Control (NIFDC). Luciferase 1000 Assay System was purchased from Promega (Madison, WI, USA).
Animals
All mice were raised in a specific pathogen-free facility, and the Institutional Animal Care and Use Committees at the Institute of Process Engineering, Chinese Academy of Sciences approved all experimental animal protocols (approval ID: IPEAECA2020401 and IPEAECA2021403). This study was performed in strict accordance with the Regulations for the Care and Use of Laboratory Animals and Guideline for Ethical Review of Animals (China, GB/T35892-2018).
Preparation of rMASE and iMASE
rMASE was prepared through layer-by-layer assembly of alum and antigen on the o/w interface. First, the alum/HA-assembled droplets were prepared by ultrasonication (Branson Digital Sonifier, total time = 120 s, power = 25%, interval time = 4 s) of the mixture of HA, alum, water, and squalene (Sigma, Germany). The alum concentration was 0.5 mg/mL, and the o/w ratio was fixed at 1/9. Then, an additional alum (0.5 mg/mL) was added into alum/HA-assembled droplets and mixed for about 30 min, and subsequently adsorbed with NP (0.05 mg/mL) to constitute the multi-layered alum-stabilized emulsion. Similarly, iMASE was prepared with NP entrapped on the inside, and HA absorbed on the outside.
As a control, an alum-stabilized emulsion was prepared by single-step sonication (total time = 120 s, power = 30%, interval time = 4 s) of alum as the colloidal stabilizers and squalene as the dispersion phase.
Interactions between outer alum and alum/HA-assembled droplets
QCM-D (Biolin Science/Q-Sense, Sweden) was used to test the interactions between the outer alum and alum/HA-assembled droplets. The chips were modified by spin-coating with alum/HA-assembled droplets, HA and alum-stabilized emulsions, respectively. Once the modified chips were installed, alum (0.5 mg/mL) was added to the QCM chamber at a continuous flow rate of 50 μL/min. The vibration frequency (ΔF) was measured to illustrate the adsorption tendency of the outer alum.
Characterization of rMASE and iMASE
To characterize the surface topography, the droplets were solidified and observed using scanning electron microscopy (SEM; JEOL, Japan). Briefly, solid alum/HA-assembled droplets were prepared using paraffin wax as the dispersion phase. The multi-layered solidified droplets were generated by cooling the temperature from 50 °C to 4 °C, and the outer alum (0.5 mg/mL) was added to attach for approximately 30 min. The droplets were observed by SEM after appropriate dilution (1:40).
Three-dimensional structured illumination microscopy (3D-SIM; GE Healthcare, Issaquah, USA) and stimulated emission depletion microscopy (STED; Leica, Mannheim, Germany) were used to track the assembly of rMASE. Briefly, the indicated droplets were prepared through the assembly of Cy3-labeled HA, Cy5-labeled NP, and Lumogallion-labeled alum on the droplets. With the diluted droplets (1:50), the assembly steps were observed in detail at ×100 magnification. The surface elemental composition of rMASE was precisely determined using inductively coupled plasma mass spectrometry (ICP-MS; Agilent Technologies, Santa Clara, CA, USA).
Coverage of the inner antigen and the surface display of outer antigen
The specific binding properties of the antigen and antibody were used to analyze whether the attached alum shielded the inner antigens. To eliminate the non-specific interactions of the antibodies, the formulations were blocked with 4% FBS. The treated droplets were then incubated with anti-HA (Sino Biological Scientific) and anti-NP antibodies (Creative Biolabs). After removing the excess antibodies, Alexa Fluor 647-coupled goat anti-Rabbit IgG (H + L) cross-adsorbed secondary antibody (Thermo Fisher Scientific) and Alexa Fluor 488-coupled goat anti-mouse IgG (H + L) cross-adsorbed secondary antibody (Thermo Fisher Scientific) were added to maintain the reaction at 4 °C for 30 min. After washing off the uncombined secondary antibodies, the droplets were observed under a confocal laser scanning microscope (CLSM; Nikon, Japan).
Force tendency of the inner and outer antigen
An X-ray diffractometer (XPert3 MRD, X-ray Stress Analyzer, Malvern, UK) was used to measure the residual stress. Data collection was performed using the side-inclination method (Ψ-goniometer). The analysis was performed with an operating voltage of 45 kV and a current of 0.8 mA. The diffraction patterns were obtained using Cu-Kα radiation of wavelength 1.54 Å and an angle from 50° to 60° in steps of 0.02° intervals in the transverse direction. The least-squares method was used to regress each data point into a straight line.
$$K = \frac}}\tan \theta _0 \bullet \frac}$$
From the above equation, we measured and calculated the modulus of elasticity E and Poisson’s ratio υ to calculate K, which was combined with the linear slope M to obtain the residual stress equation:
Thermal stability was estimated using differential scanning calorimetry (DSC; Netzsch, Germany) to assess the antigens release trend. An empty alum pan was used as the reference. The heating rate of the samples was 10 °C/min in the temperature range of 25–200 °C. The locations of the thermal peaks were determined using GraphPad Prism 9.
Evaluation of antigen release
To analyze the intracellular alum dynamics, BMDCs were co-incubated with rMASE or iMASE for different periods. In the following step, cells were fixed with 2.5% glutaraldehyde overnight in a 0.5 M phosphate solution. Afterward, cells were postfixed in osmium tetroxide, dehydrated in ethanol, and embedded in Epon. To observe the cells, the dishes were broken into pieces and glued onto Epon sticks, and a range of 80–100 nm was selected for each section. Uranyl acetate and lead citrate were used to stain slices. Transmission electron microscopy (TEM; JEOL, Japan) was performed to obtain images at ×10,000 magnification.
The intracellular antigen release profile was evaluated using a high-content live-cell imaging system (Operetta CLS, PerkinElmer, Waltham, MA, USA). rMASE and iMASE were prepared using FITC-HA and Cy5-NP, respectively, and were co-incubated with BMDCs for 6 h to allow maximum uptake. Residual droplets were removed from the medium. Subsequently, the fluorescence intensities of HA and NP were measured at the indicated times.
Antigens depot and uptake
To measure antigen retention at the injection site, BALB/c mice (6–8 weeks, female) were intramuscularly administrated the indicated formulations containing 5 µg/dose Cy5-HA conjugate and 5 µg/dose Cy7-NP conjugate (Thermo Fisher). The in vivo imaging system FX Pro (Kodak, Rochester, USA) was used to collect fluorescence signals of the antigens at the injection site after intramuscular administration at the indicated time points.
In addition, we evaluated the uptake in vivo by the intramuscular administration of the indicated formulations to BALB/c mice (6–8 weeks, female). Specifically, muscle tissues at the injection sites were harvested and digested with 0.2% (w/v) collagenase type II for 2 h at 37 °C. Afterward, the cells were collected and blocked with anti-mouse CD16/CD32 antibody. After washing thrice, the cells were stained with Ghost Dye™ Violet 450 Viability, PE dump channel markers (anti-mouse F4/80 and Ly-6C antibodies), and PerCP-Cy5.5 anti-mouse CD11c antibody. Data were collected using flow cytometry (CytoFlex LX; Beckman Coulter).
Antigen-specific antibody secretion
BALB/c mice (6–8 weeks, female) were intramuscularly administered the indicated vaccines, and serum was collected to detect antigen-specific antibody titers. Briefly, 100 μL of antigen diluent (2 μg/mL) was added to 96-well plates and stored at 4 °C overnight. On the second day, after washing with PBS–0.5% Tween 20 (PBST) three times, the 96-well plates were blocked with 0.5% bovine serum albumin (BSA) at 37 °C for 1 h. The serum was diluted to the appropriate dilution in the plates and incubated at 37 °C for 40 min. Afterward, the HRP-conjugated goat anti-mouse IgG antibody (1:50000; Abcam) was added to react at 37 °C for another 40 min. After washing six times, TMB substrate (50 μL per well) was added, and the reaction was immediately stopped. OD450 values were measured using an Infinite 200 PRO (TECAN, Mannedorf, Switzerland). Antibody titers were defined as the end-point of dilutions where the OD450 value was greater than or equal to twice that of the negative control group. Antigen-specific IgG1 and IgG2a titers were detected on day 28 using the same method.
ELISPOT evaluations
For ELISPOT evaluation, polyvinylidene difluoride (PVDF)-based membrane plates (Millipore) were activated with 35% ethyl alcohol. Then the plates were coated with anti-mouse IFN-γ or IL-4 antibodies (5 μg/mL) and stored at 4 °C overnight. The next day, the plates were blocked with the culture medium (RPMI 1640, 10% FBS) for more than 1 h. Splenocytes from vaccinated mice were immediately added to the wells at 5 × 105 cells/well and re-stimulated with a specific antigen (2 μg/mL). On the third day, the plates were incubated with the detection antibody (1 μg/mL) for 2 h after removing the cells, followed by incubation with the streptavidin-ALP detection antibody (1 μg/mL) at room temperature. BCIP/NBT was then added to the plates and incubated for approximately 10 min in the dark. Each of the above steps requires washing the plates 3–6 times. Finally, the plates were washed with distilled water at the appropriate time points and air-dried overnight in the dark. The spots were scanned and counted using an ELISPOT Analyzer (AT-Spot 2100, China). Each experiment was repeated three times, and similar results were obtained.
Influenza A challenge experiments
For the prophylactic protection assay, BALB/c mice (6–8 weeks, female) were randomly divided into different groups and immunized twice by intramuscular injection. After 14 d, the mice were challenged intranasally with influenza A (A/Puerto Rico/8/1934) at a dose of 2 LD50 in 50 μL of PBS. The body weight and survival rate of all mice were monitored daily for 21 d. Mice were euthanized when their body weight loss exceeded 20% of their pre-challenge weight.
BALB/c mice (6–8 weeks, female) were euthanized on day 9 post-challenge to dissect the lung tissues for histology and viral load detection. To measure and quantify viral loads, a section of the lung tissue was homogenized and resuspended in 1 mL TRIzol reagent to extract total RNA for RT-qPCR analysis. The primer pair for the mRNA of the influenza virus was F-5′ AAGACCAATCCTGTCACCTCTGA-3′, R-5′-CAAAGCGTCTACGCTGCAGTCC-3′, and that for the internal reference gene (GAPDH) was F-5′-CAATGTGTCCGTCGTGGATCT-3′, R-5′-GT CCTCAGTGTAG CCCAAGATG-3′.
Evaluation of the transcriptome
The total RNA of BMDCs co-cultured with iMASE or rMASE for 24 h was extracted using the TRIzol reagent (Invitrogen). RNA libraries were sequenced on an Illumina sequencing platform (Gene Denovo Biotechnology, Co., Ltd., Guangzhou, China). In order to normalize and standardize the expression level of each gene, each part of the transcript per kilobase of transcript per million mapped reads (FPKM) was used. Differentially expressed genes (DEGs) were filtered using FPKM based on the edgeR’s general filtering criteria (log2| fold change | >1, false discovery rate [FDR] < 0.05).
DC activation at the injection sites
To evaluate DC activation, BALB/c mice (6–8 weeks, female) were intramuscularly administrated with the indicated formulations and euthanized 1-, 3-, 5-, and 7-d post-administration. The tissues at the injection site were selected and immersed in collagenase type II (0.2%, w/v) for 2 h at 37 °C, allowing for the tissue digestion and isolation of the single-cell suspension. Then, the cells were blocked with anti-mouse CD16/CD32 antibody for 20 min and stained with Ghost Dye™ Violet 450, PE-labeled dump channel markers (anti-mouse F4/80 and Ly-6C antibodies), PerCP-Cyanine5.5 anti-mouse CD11c antibody, APC anti-mouse CD40 antibody, and FITC anti-mouse CD86 antibody at 4 °C for 30 min and then analyzed by flow cytometry. Furthermore, the muscle tissues were removed from the injection sites to test the expression of CCR7 among the recruited DCs. The tissues were then transferred to a grinder and homogenized on ice. Afterward, the cells were collected and blocked with anti-mouse CD16/CD32 (mouse Fc block) antibody at 4 °C for 20 min. After washing three times, the cells were stained with Ghost Dye™ Violet 450 Viability Dye, PE dump channel markers (anti-mouse F4/80 and Ly-6C antibodies), and Brilliant Violet 605™ anti-mouse CCR7 antibody at 4 °C for approximately 30 min and then analyzed by flow cytometry (CytoFlex LX, Beckman Coulter).
To determine cytokine secretion at the injection site, approximately 80 mg of the muscle tissue was ground. After removing cell fragments, the supernatant was collected by centrifugation at 500 × g for 5 min. The cytokine levels of IFN-α, IFN-β, TNF-α, and IL-2 in the supernatants were determined using enzyme-linked immunosorbent assay (ELISA).
DC and Germinal center activations in draining lymph node
To analyze the dLN activation, BALB/c mice (6–8 weeks, female) were immunized via intramuscular injection in the calf muscles of the hind limb with 100 μL vaccine that contained 5 µg/dose RBD and 5 μg/dose NP and euthanized at the indicated days. Afterward, the lymph nodes in the popliteal fossa between the biceps femoris and semitendinosus were picked and passed through a 70 μm Cell Strainer (BD Falcon) to obtain single-cell suspensions. In terms of DC subsets in dLN, the obtained single cells were blocked with anti-mouse CD16/CD32 antibody and stained with Ghost DyeTM Red 780, PerCP-Cyanine5.5 anti-mouse CD11c antibody, FITC anti-mouse CD8α antibody, PE anti-mouse CD11b antibody, and PE-Cy7 anti-mouse CD103 antibody. Data were collected by flow cytometry. To detect DC activation, obtained single cells were blocked with anti-mouse CD16/CD32 antibody and stained with Ghost Dye™ Violet 450 Viability, FITC anti-mouse CD11c antibody, and APC anti-mouse CD40 antibody at 4 °C for approximately 30 min, and then analyzed through flow cytometry. In addition, CD40L+ T-cells were tested. Briefly, cells were labeled with APC-Cy7 anti-mouse CD3e and eFluor 450 anti-mouse CD40L antibodies and analyzed by flow cytometry.
Tfh and GC B cells were then probed. Briefly, mice were injected intramuscularly with the indicated vaccines and euthanized 7 d post-administration. Afterward, the LN cells were prepared and labeled with the Ghost Dye™ Violet 450, PE dump channel markers (anti-mouse B220, CD11b, CD11c, and F4/80 antibodies) and antibodies against Tfh (PerCP-Cyanine5.5 anti-mouse CD3e, APC-Cy7 anti-mouse CXCR5, and PE-Cy7 anti-mouse ICOS antibodies). To confirm the presence of GC B cells, cells were stained with Ghost Dye™ Violet 450, PE dump channel markers (anti-mouse CD3, CD11c, CD11b, and F4/80 antibodies), and antibodies against GC B cells (FITC anti-human/mouse B220, eFluorTM 660 anti-mouse GL-7, and PerCP-eFlour 710 anti-mouse CD95 antibodies). Antibody dilution was performed for flow cytometry staining according to the manufacturer’s handbook. We defined GC B cells as FAS+ GL-7+ B220+ and Tfh cells as CXCR5+ ICOS+ CD3+.39,40 Data were collected by flow cytometry.
Immunofluorescence staining was performed as previously described.41 LNs were fixed with paraformaldehyde, embedded in paraffin, and sectioned into slices of 5–6 μm thickness. After deparaffinization and hydration, the samples were washed with Tris-buffered saline containing Tween® 20, treated with 3% H2O2 for 10 min, and blocked with 2% BSA solution for 30 min. The desired concentration of primary antibody was diluted in 500 µL of 0.1% BSA (CD4, CXCR5, or ICOS), added to the cells, and incubated overnight at 4 °C. The corresponding secondary antibodies were incubated after washing at 1:500 in 2% BSA blocking solution for 1 h at room temperature. After washing, the slides were incubated with 4′-6-diamidino-2-phenylindole in PBS for 5 min. Finally, the autofluorescence was quenched, and the slides were detected using K-Viewer.
Afterward, LN-residing memory B cells were evaluated. Briefly, LNs were collected two weeks after boost immunization. The cells were then stained with Ghost Dye™ Violet 450 Viability, PE dump channel markers (anti-mouse CD3e, CD11c, CD11b, and F4/80 antibodies), FITC anti-mouse B220 antibody, and APC anti-mouse CD27 antibody. Data were collected using flow cytometry.
Binding affinity measurement
To test antibody affinity, serums from immunized mice were purified using protein A antibody affinity chromatography. The binding affinity of antibodies to the RBD antigen was tested by BLI using the Octet® R8 system (Startorius BioAnalytical Instruments Inc.). Briefly, the biotinylated RBD antigen was diluted to 1200 μL and cured for 600 s. Afterward, the RBD antigen was diluted with PBST to 200 nM and then half-diluted for a total of six concentration gradients (200 nM, 100 nM, 50 nM, 25 nM, 12.5 nM, and 6.25 nM). Antibody samples were added to a 96-well plate, and the assay program was set. Ligand-loaded Streptavidin (SA) biosensors were then incubated with different concentrations of RBD antigen in a kinetics buffer. A global fit of the binding curves generated the best fit with the 1:1 model, and kinetic association and dissociation constants were determined. The data were aligned to obtain the kinetic parameters (KD) using Fortebio data analysis 12.0.
Pseudovirus neutralization assay
A published method was used to evaluate the neutralization of SARS-CoV-2. First, the TCID50 was determined by infection of Huh7 cells. Afterward, serial dilutions of heat-inactivated serum collected from immunized mice were incubated with 100 TCID50 of pseudovirus at 37 °C for 1 h. The mixture was then added to 96-well plates that contained Huh7 and incubated for 24 h. Finally, the cells were lysed, and a Luciferase Assay System (Promega, USA) was applied to evaluate luciferase activity. Experiments were performed according to the manufacturer’s instructions. A relative light unit (RLU) reduction greater than 90% compared with the virus control well was defined as NT90, the highest reciprocal serum dilution at which RLUs were reduced. To determine half of the limit of detection, an NT90 below that level was considered.
Detection of memory T cells
BALB/c mice (6–8 weeks, female) were euthanized 28 d after the first immunization, and splenocytes were isolated and gently passed through a 100 μm cell strainer (BD Falcon). Next, erythrocyte lysis buffer was added, followed by washing three times with RPMI 1640 to obtain single-cell suspensions. Then, the single-cell suspensions (4 × 106 cells/well) were re-stimulated with the antigen (2 μg/mL) at 37 °C in a humidified, 5% CO2 atmosphere. After 48 h of culture, the splenocytes were collected and stained with memory T cell related-fluorescent antibodies, including Ghost Dye™ Violet 450, PE dump channel markers (anti-mouse B220, CD11c, CD11b, F4/80, and Ly-6C antibodies), PE-Cy7 anti-mouse CD3e antibody, APC-Cy7 anti-mouse CD4 antibody, PerCP-Cyanine5.5 anti-mouse CD8a antibody, APC anti-mouse CD44 antibody, and FITC anti-mouse CD62L antibodies. CD44high CD62Lhigh and CD44high CD62Llow are known as the central memory T cells and effector memory T cells, respectively. Data were collected by flow cytometry.
SARS-CoV-2 challenge experiments
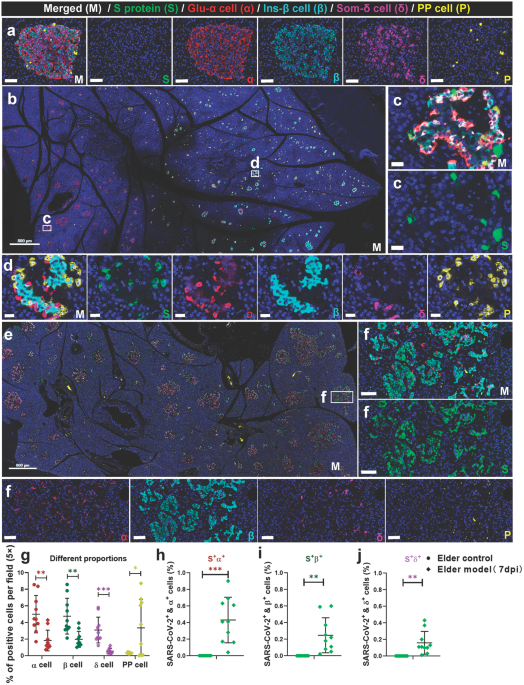
For SARS-CoV-2 challenge experiments, BALB/c mice (6–8 weeks, female) were vaccinated with iMASE and rMASE via intramuscular injection on days 28 and 49 before the challenge and compared with the non-immunized mice (term “Sham”). Challenge experiments and immunoassay were performed on the same day under consistent conditions for all the groups. Briefly, mice were intranasally transduced with 8 × 108 vp of Ad5-hACE2 23 or 44 days after the first immunization with iMASE and rMASE to rapidly induce the mouse model of SARS-CoV-2 infection. Five days later, the transduced mice were given 5 × 105 TCID50 of SARS-CoV-2 (hCoV-19/China/CAS-B001/2020; GISAID No. EPI_ISL_514256-7) via the intranasal administration,42 and all mice were euthanized and necropsied 3 d after challenge. Finally, lung tissues were harvested to calculate the viral loads and detect the pathological status. All animal experiments with SARS-CoV-2 the challenge were operated at the Animal Biosafety Level 3 (ABSL3) facility of IMCAS.
Determination of viral loads in the lung
For the determination of viral loads in lung tissue samples, RT-qPCR was used. First, the lung tissues of mice were collected and homogenized. Following homogenization and centrifugation, 50 μL of the supernatant was used to extract RNA from SARS-CoV-2 using a MagMAXTM Express Magnetic Particle Processor (Applied Biosystems, USA). RT-qPCR assays were conducted using the FastKing One Step Probe RT-qPCR kit (Tiangen Biotech, China), according to the manufacturer’s instructions.43 The N gene of the SARS-CoV-2 genome was detected using the corresponding primers and probes.
Histopathology analysis
Three days after infection, necropsies were performed on six mice per group following a standard protocol. We collected the lungs of challenged mice, fixed them in 10% neutral buffered formaldehyde, and embedded them in paraffin. Tissue sections (5 μm) were stained with H&E to reflect the characteristics of infection, such as interstitial pneumonitis and alveolitis.
Live SARS-CoV-2 neutralization assay
For live SARS-CoV-2 neutralization assay, we inactivated the plasma samples collected from mice at 56 °C for 0.5 h. Following serial dilution with cell culture medium at a concentration of 1: 4 or 50,000 ng/mL, the inactivated diluted serum was mixed with 100 TCID50 of the SARS-CoV-2 virus and incubated at 37 °C for 1 h. Next, the mixtures were added to the 96-well plates covered with confluent Vero cells. Next, the mixture was incubated for another 5 d at 37 °C. Three different individuals were observed, and the cytotoxic effect (CPE) of each well was recorded under a microscope. The CPE was then used to calculate neutralizing titers using the Reed Muench method. All experiments were conducted at the biosafety level 3 (BSL3) facility of the SINOVAC.
Statistics
All animal studies were performed after randomization. All values are expressed as the mean ± the standard error of the mean (s.e.m). Data were analyzed by one- or two-way analysis of variance (ANOVA) for comparison of multiple groups using GraphPad Prism 9 and Origin 9 software. Flow cytometry data were analyzed using FlowJo 10.0 and CytExpert Software 2.3. Statistical significance was set at a P-value less than 0.05 (P < 0.05).



留言 (0)