
記住我


Chromatin is the DNA and histone protein macromolecular complex that supplies the scaffold for the packaging of our whole genome. It contains the genetic material of eukaryotic cells. The fundamental functional unit of chromatin is the nucleosome. It is composed of 147 DNA base pairs wrapped around an octamer of histones H2A, H2B, H3, and H4. All of the nucleosome components are susceptible to covalent alteration, which significantly modifies the structure and function of these key chromatin constituents, as revealed by research into the coordinated control of the nucleosome.
The term epigenetics was coined by Conrad Waddington to describe the process by which modifications to a cell’s phenotype can be passed down across generations without requiring a change to the DNA sequence. A consensus definition of epigenetics is lacking, and the definitions remain vague after decades of debate and research.1 Therefore, the word epigenetics will be used throughout this review to refer to chromatin-based activities that govern DNA-programmed processes.
Chromatin-modifying enzymes actively add to and remove modifications from DNA and histones in a highly controlled way. Currently, at least four different modifications to DNA and histones have been identified.2,3 These alterations can alter the structure of chromatin by modifying the noncovalent interactions between and within nucleosomes. In addition, they serve as docking sites for proteins with specific domains that may identify these alterations. These chromatin readers recruit other chromatin modifiers and remodeling enzymes to implement the changes.
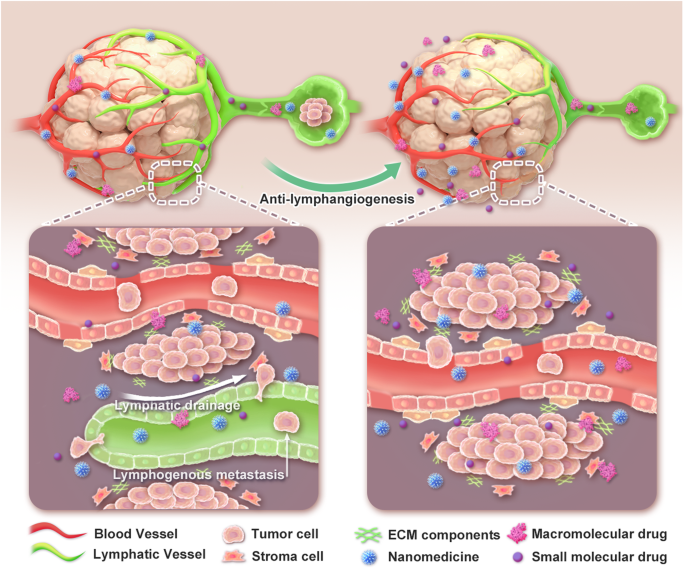
Numerous oncogenes and tumor suppressor genes can accumulate mutations and epigenetic modifications that lead to cancer.4,5 Increasing evidence suggests that epigenetic alteration is involved in a number of tumor cell biological activities, such as proliferation, invasion, metastasis, and metabolic reprogramming.6,7 Malignant cell differentiation, proliferation, invasion, metastasis, and even medication therapy resistance is influenced by the interplay between malignant cells and the immediate environment influences, affecting how a tumor progresses.6,8 Recent research has demonstrated that epigenetics regulates immune cell activation and infiltration into TME, which may alter immunotherapy efficacy.9,10 Therefore, epigenetic alterations are potential tumor immunotherapy targets that can be employed in conjunction with treatments such as immune checkpoint inhibitors (ICIs) to significantly improve tumor patient survival and quality of life. Here, we present a current and comprehensive summary of epigenetic modification and associated immunological responses in the TME. In addition, the possibility of targeting epigenetic regulators in cancer immunotherapy is highlighted.11
Epigenetic modificationsDNA methylationThe attachment of a methyl group to the 5-carbon of cytosine (5mC) in CpG dinucleotides was the first identified type of epigenetic alteration and is the most well-studied modification of chromatin.12,13 Telomeres, dormant X chromosomes, centromeres, and repetitive DNA sequences are common sites of DNA methylation.14 DNA methylation is involved in a wide variety of biological processes, such as X-chromosome inactivation, imprinting, and the maintenance of genomic stability.15,16,17,18 In cancer, global DNA hypomethylation was first observed experimentally ~30 years ago.19 Global DNA hypomethylation and hypermethylation of tumor suppressor gene promoters are hallmarks of cancer cells and key driver of carcinogenesis.20
DNA methylation is a dynamic process that can be influenced by writers, erasers, and readers. The DNA methyltransferase (DNMT) enzymes DNMT1, DNMT3A, and DNMT3B transfer a methyl group from S-adenosyl-l-methionine to the cytosine residue. DNMT1 is a maintenance methyltransferase that detects hemimethylated DNA created during DNA replication and methylates newly synthesized CpG dinucleotides whose parental strand partners are already methylated.21 DNMT3A and DNMT3B, despite their ability to methylate hemimethylated DNA, largely function as de novo methyltransferases to initiate DNA methylation during embryogenesis.22
5mC can be demethylated to 5-hydroxymethylcytosine (5hmC) via erasers. Indeed, 5hmC is iteratively oxidized to produce additional oxidative derivatives, including 5-formylcytosine (5fC) and 5-carboxycytosine (5caC). Iterative oxidation reactions are performed by the ten-eleven translocation (TET) family of proteins (Fig. 1). In mammalian DNA, the TET1–3 protein family is responsible for the catalytic conversion of 5mC into 5hmC. In addition, various oxidation derivatives, including 5fC and 5caC, are produced through the repeated oxidation of 5hmC by the TET family members.6 5hmC is involved in transcriptional activation and inhibition, and TET proteins have been identified as having common activities.3
Fig. 1
DNA methylation. DNA methylation is a dynamic process modulated by writers, erasers and readers. DNA methyltransferases (DNMTs) enzymes (“writers”) transfer a methyl group from S-adenosyl-L-methionine to the cytosine residue (5mC), including DNMT1, DNMT3A and DNMT3B. 5mC can be demethylated to 5-hydroxymethylcytosine (5hmC) via erasers. Indeed, 5hmC is iteratively oxidized to produce further oxidative derivatives, including 5-formylcytosine (5fC) and 5-carboxycytosine (5caC). The iterative oxidation reactions are performed by the ten-eleven translocation (TET1–3) family of proteins
To coordinate these downstream regulatory processes, DNA methylation provides a platform for various methyl-binding proteins (“readers”), which regulate the crosstalk between DNA methylation, histone modifications, and chromatin architecture. MeCP2, the prototypical member of the methyl-CpG-binding domain (MBD) family of proteins (MBD1, MBD2, and MBD3), recruits histone-modifying enzymes, chromatin remodelers, and DNMTs to methylated CpGs involved in gene repression.23,24,25,26,27
Histone modificationHistone modifications are a class of post-translational modifications (PTMs) that impact chromatin structure and have been found to have a crucial influence on transcription and all DNA-template processes. Typically, marks are located on the N-terminal ‘tails’ of histones and contribute to nucleosome stability.28 At least 16 distinct histone PTMs, including acetylation, methylation, and phosphorylation, have been discovered to date. Histone modifications are dynamically regulated by proteins called “writers”, “readers”, and “erasers”. The transcriptional activation or repression of genes is affected by abnormal histone modifications, which also influence numerous processes, including DNA replication and recombination, and hence impair cell homeostasis and control tumor formation.6,29
Histone acetylationEnzymes called histone acetyltransferases (HATs) add acetyl groups to the ε-amino group of lysine side chains. Acetyl-CoA is a cofactor for a number of enzymes with roles in transcription, chromatin structure, and DNA repair.30 The neutralization of lysine’s positive charge by acetylation may impair the electrostatic connection between positively charged histones and negatively charged DNA. Consequently, histone acetylation is frequently linked to a more “open” chromatin conformation.
Type-B HATs are primarily cytoplasmic; they acetylate free histones but not those already deposited into chromatin, and type-A HATs are primarily nuclear; they are divided into three subfamilies based on amino acid sequence homology and conformational structure: the Gcn5-related N-acetyltransferase family (GNATs), the MYST family (MOZ, Ybf2, Sas2, and TIP60), and the orphan family (CBP/EP300 and nuclear receptors).31,32
Histone deacetylases (HDACs) counteract the effects of HATs and restore the positive charge of the lysine side chains. HDACs are substrate-specific, meaning they can target not only histones but also nonhistone proteins such as HATs. HDACs are classified into four primary groups: class I (HDAC1, HDAC2, HDAC3, HDAC8), class IIa (HDAC4, HDAC5, HDAC7, HDAC9), class IIb (HDAC6, HDAC10), class III (sirtuin1-7), and class IV (HDAC11) HDACs. Only class III HDACs require nicotinamide adenine dinucleotide(NAD), while classes I, II, and IV HDACs require Zn2+.33
In addition, acetylation can serve as a signal in chromatin that is recognized by “readers” (a subset of bromodomain proteins called bromodomain and extraterminal domain (BET)). The BET family comprises of four members with a common architecture and structural design: BRD2, BRD3, BRDT, and BRDT. Targeting the BET bromodomains with epigenetic-based drugs is thus likely to be a potential cancer strategy. In addition, BET proteins are involved in a number of essential processes, including transcription initiation, transcription elongation, cell-cycle progression, DNA damage regulation, and telomere regulation.34,35,36,37
Histone methylationOn the side chains of lysine, arginine, and histidine residues, histones can be methylated without changing the total charge of the molecule. Monomethylated, dimethylated, and di-asymmetrically methylated forms of arginine can exist, as can monomethylated, dimethylated, and trimethylated forms of lysine. Among these types of methylation, histone lysine methylation has received the most attention. SUV39H1 was the first histone lysine methyltransferase (HKMT) targeting histone 3 lysine 9 (H3K9) to be found.38 HKMTs catalyze the transfer of a methyl group from S-adenosine methionine (SAM) to the ε-amino group of lysine. Remarkably, except for the Dot1 enzyme methylating H3K79, all HKMTs that methylate N-terminal lysine possess an enzymatically active SET domain. Histone lysine methyltransferases (HKMTs) are relatively specific. Histone 3 lysine 9 (H3K9) can be trimethylated (H3K9me3) from a monomethylated (H3K9me1) state by KMT1A/B, or it can be methylated to a dimethylated (H3K9me2) form by the H3K9 methyltransferase KMT1C (also known as G9a), with a preference for monomethylation to dimethylation.39,40 Different methylation sites have different effects. Examples of sites of histone modifications that are associated with euchromatin activity include H3K4, H3K36, and H3K79, while sites of histone modifications such as H3K9, H4K20, and H3K27 are associated with heterochromatin.41 Diverse methylation statuses on the same residue also have different functional implications. For example, trimethylation of H3K9 is associated with transcriptional repression, whereas monomethylation of H3K9 is present in actively transcribed genes.
In 2004, lysine-specific demethylase 1 (LSD1) was discovered to use FAD as a cofactor to reverse lysine methylation.42 JMJD2 was the first identified tri-methyl lysine demethylase with a distinct catalytic mechanism distinct from that of LSD1, employing Fe (II), alpha-ketoglutaric acid, and a free radical attack mechanism.43 JMJD2 demethylates H3K9me3 and H3K36me3. Demethylases, similar to histone methyltransferases, have a high substrate selectivity. They are also sensitive to the degree of lysine methylation; for example, certain enzymes can only demethylate monomethylated and dimethylated substrates, whereas others can demethylate all three forms of methylated lysine.
Histone phosphorylationSimilar to histone acetylation, the phosphorylation of histones is a highly dynamic process that is reciprocally regulated by protein kinases and protein phosphatases. It primarily, but not entirely affects serines, threonines, and tyrosines in the N-terminal tails of histones. Protein kinases and phosphatases, which add and remove the modification, respectively, work together to control the overall level of the modification.44 In general, histone phosphorylation sites are related to transcriptional regulation and in chromatin condensation.45 Most phosphorylation sites on histones are located in the N-terminal tails. However, there are sites within the main regions. One such instance is the phosphorylation of H3Y41 mediated by the nonreceptor JAK2.46
Except for the above three histone modifications, there are a variety of less prevalent and atypical PTMs, such as histone ubiquitination, ADP-ribosylation, deamination, and O-GlcNAcylation, etc., which have been reviewed in detail in refs. 47,48
RNA modification N6-methyladenosineThe m6A modification appears mostly on the common sequence 5’-RRACH-3’ (R = A or G and H = A, C, or U),49,50,51 and the modification is mainly localized near a stop codon in a 3′ untranslated regions (3’UTRs) within a lengthy internal exons.52,53,54 Dynamic and reversible, the m6A modification process is mediated by m6A methyltransferases (writers), m6A demethylases (erasers), and m6A-binding proteins (readers).55
The dynamic process involved in the m6A deposition is regulated by a methyltransferase complex (writer). As the first known m6A methyltransferase and a major catalytic subunit, methyltransferase-like 3 (METTL3) binds S-adenosylmethionine (SAM) and transfers methyl groups from SAM to adenine bases in RNA.49,56,57 Methyltransferase-like 14 (METTL14) is necessary for the identification of RNA substrates and forms a stable heterodimer with METTL3, hence increasing the complex’s catalytic activity.51,58,59 As the main regulatory component of the complex, Wilms’ tumor 1-associating protein (WTAP) participates in the localization of METTL3-METTL14 heterodimers to nuclear speckles, thereby facilitating m6A modification.60 Furthermore, the complex includes zinc-finger CCCH domain-containing protein 13 (ZC3H13), RNA-binding motif protein 15 (RBM15), KIAA1429 (also called VIRMA), and its paralog RBM15B. It has been reported that KIAA1429 recruits and guides the localization of m6A methylation to the 3’ UTRs and close to a stop codon.58,61 ZC3H13 is a novel cofactor that binds with other components (such as RBM15 and WTAP) to regulate nuclear m6A modification.62 RBM15/15B is also crucial for the recruitment of writers to target sites.63,64 Recently, studies have shown that ZCCHC4,65 METTL566,67 and METTL1668 can function as methyltransferases and thus contribute to the m6A modification of some small nuclear RNAs (snRNAs), noncoding RNAs (ncRNAs) and pre-mRNAs.
Demethylases (erasers) remove methyl groups from N6 adenosine. Two primary erasers are fat mass and obesity-associated protein (FTO)69 and alpha-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5).70 These two proteins both eliminate the m6A mark from RNA to reverse the m6A modification. Moreover, alkB homolog 3 (ALKBH3) has been shown to enhance protein synthesis in cancer cells by mediating tRNA demethylation.
The m6A-binding proteins (readers) recognize and interact with the m6A marks on target transcripts.71 Different readers can drive multiple biological processes, such as mRNA splicing, export and stability, miRNA biogenesis, translation efficiency, and RNA structure switching. The YTH domains include the YTH domain family proteins 1, 2, and 3 (YTHDF1, YTHDF272,73, and YTHDF374,75) and YTH domain-containing proteins 1 and 2 (YTHDC176,77 and YTHDC278). In addition, insulin-like growth factors (IGF2BP1-3) are critical for enhancing mRNA stability.79 Furthermore, other readers, such as heterogeneous nuclear ribonuclease (HNRNP) family members (HNRNPA2B1,80,81 HNRNPC, HNRNPG,82 eukaryotic translation initiation factor 3 (eIF3), and fragile X mental retardation protein (FMRP), have also been demonstrated to perform a range of biological functions83 (Fig. 2).
Fig. 2
Internal RNA modifications. The main players in the deposition, removal and downstream recognition of the modification are listed together with the effect of modifications on base pairing. ADAR adenosine deaminase acting on double-stranded RNA, ADAT adenosine deaminase acting on transfer RNA, ALKBH alkB homolog, CTU cytoplasmic transfer RNA 2-thiolation protein, DKC1 dyskerin pseudouridine synthase 1, DNMT2 DNA methyltransferase-like2, ELP elongator complex protein, FTO fat mass and obesity-associated protein, m1A N1-methyladenosine, m6A N6-methyladenosine, m5C 5-methylcytosine, m7G 7-methylguanosine, METTL methyltransferase-like, NSUN NOL1/NOP2/SUN domain family member, PUS pseudouridine synthase, RNMT RNA guanine-7 methyltransferase, RPUSD RNA pseudouridine synthase domain-containing protein, TRM6 transfer RNA methyltransferase non-catalytic subunit 6, TRM61 transfer RNA methyltransferase catalytic subunit 61, TRMT10 transfer RNA methyltransferase 10, tRNA transfer RNA, WBSCR22 Williams–Beuren syndrome chromosomal region 22 protein, YTHDC YTH domain-containing, YTHDF YTH domain-containing family, ZCCHC4 zinc-finger CCHC domain-containing protein 4, NAT10 N-acetyltransferase 10
N1-methyladenosineIn contrast to m6A marks, the N1-methyladenosine (m1A) marks show significantly lower abundance in mammalian tissues. Recent in-depth studies have shown that m1A modification is widely distributed throughout the whole transcriptome. Through electrostatic effects, the m1A mark can modulate RNA secondary structures and RNA‒protein interactions. m1A is enriched at translation start sites of mRNAs (5’ UTR) and in multiple regions of tRNAs, where it can upregulate translation and mediate a variety of corresponding biological processes.84,85,86
The tRNA methyltransferase catalytic subunit 6 (TRM6)–tRNA methyltransferase catalytic subunit 61 (TRM61) complex (TRM6–TRM61) is the only known methyltransferase capable of catalyzing the addition of N1-adenosine in mRNAs.87 Moreover, this complex and tRNA methyltransferase 10 homolog A (TRM10) can both direct the m1A modification of tRNAs.
AlkB homolog 3 (ALKBH3) is critical for removing the methyl group from m1A modification in mRNAs.88 In tRNAs, alkB homolog 1 (ALKBH1) 89 and ALKBH390 can both modulate tumor progression by catalyzing m1A modification. Biochemical assays have indicated that YTHDF2 binds to m1A; therefore, YTHDF2 may be a potential m1A “reader”, a supposition that needs to be further verified.91 Recently, Zheng et al. identified YTHDF3 as the m1A “reader” by mass spectrometry92 (Fig. 2).
5-Methylcytosine5-Methylcytosine (m5C) carries a methyl group at the cytosine C5 position. The known writers to date include DNA methyltransferase 2 (DNMT2) and NOP2/Sun domain family members 1-7 (NSUN1-7).93,94,95 As the most researched methyltransferase, NSUN2 catalyzes the m5C modification of mRNAs, tRNAs, rRNAs, mitochondrial tRNAs (mt-tRNAs), long ncRNAs (lncRNAs), ncRNAs, and other RNAs.96,97,98,99,
留言 (0)