
記住我
T cells play a central role in cancer immunosurveillance and eradication.1 The generation of effective tumor-directed T cell responses requires many steps such as (1) the activation of effector T cell function, (2) formation of effector memory T cells, and (3) activation of an intrinsic capacity to expand and infiltrate solid tumors while remaining functional despite the tumor microenvironment (TME). Across these steps, one of the fundamental adaptable biological programs supporting T cells is the molecular machinery responsible for antigen receptor signaling.2 Intracellular signals encoded by T cell receptor (TCR) engagement can quantitatively discriminate between antigens of differing affinities. As a counterpart to these positive signaling pathways, negative signaling loops are critical to maintain a T cell activation threshold.3
Our understanding of the balance between stimulatory and inhibitory signals necessary for effective immune responses is constantly evolving and could be used to develop immunotherapy strategies. Indeed, the dynamic interplay between inhibitory and stimulatory signals on T cells modulates the degree of immune activation to allow tolerance to self-antigens (inhibitory) while mounting an adaptive immune response to foreign antigens (stimulatory).4 An essential mechanism of inhibitory stimuli coming from immune checkpoints (ICPs) expressed at the surface of T cells (such as PD-1/PD-L1 pathway and CTLA4) is to control the inflammatory response and to protect normal cells from T cell–mediated cytotoxicity after their activation. T cell exhaustion is mediated by the upregulation of ICPs.5 By blocking the checkpoint engagement, immune checkpoint inhibitors (ICIs) prevent T cell exhaustion.5 6 These ICIs are currently used to treat cancer. Like ICPs, some intracellular proteins are involved in negative feedback loops downstream the TCR. Recently, they have been considered as potent targets in the context of cancer immunotherapies.
Recently, our team and others demonstrated the capacity to target TCR signaling inhibitory intracellular proteins (intracellular immune checkpoints (iICPs)) to enhance T cell–based immunotherapies.7–10 Here, we highlight numerous negative feedback mechanisms of TCR signaling with a potential to improve cytotoxic T cell function during immunotherapy. Some strategies to block these iICPs in clinical development as cancer immunotherapies. Indeed, the regulatory mechanisms of TCR-mediated signaling vary in different T cell maturation or differentiation states, as well as differences between conventional TCR and chimeric antigen receptor (CAR)-T cell signalosomes.11 The complexity of these regulatory loops of TCR activation needs to be carefully studied when considering possible therapeutic approaches.
Due to their intracellular localization, targeting iICPs remains challenging. Nonetheless, pharmacological approaches based on systemic administration of small molecules have already been reported12 (ie, clinical trials NCT04521413, NCT04649385, NCT05128487, NCT05159700, NCT05233436, NCT05370755, NCT05107674, NCT05107739, NCT05662397, NCT05315167). The recent development of PROTACs (Proteolysis Targeting Chimeras) has expanded the toolbox of chemicals available,10 especially when considering targeting ‘undruggable’ proteins, that is, without enzymatic activities. Indeed, in vitro gene editing coupled with adoptive cell therapy allows T-cell-specific deletion of iICPs in clinically relevant settings (ie, clinical trials NCT04426669, NCT05566223). Hopefully, future improvements in gene therapies, particularly in delivery, will enable in vivo gene modification.13 Therefore, combination of both fundamental knowledge of TCR signaling regulation and cutting-edge technologies may open a new era in immunotherapy.
TCR signaling modulesThe TCR determines lymphocyte T activation, differentiation and fate.2 TCR signaling is characterized by a complex structure of protein-protein interactions which define the response of T cells by acquisition of phenotypic, genomic and functional modifications. TCR signaling response is defined as a two-step process represented by two modules of protein associations: initiation and amplification of the TCR encoding signals (figure 1).
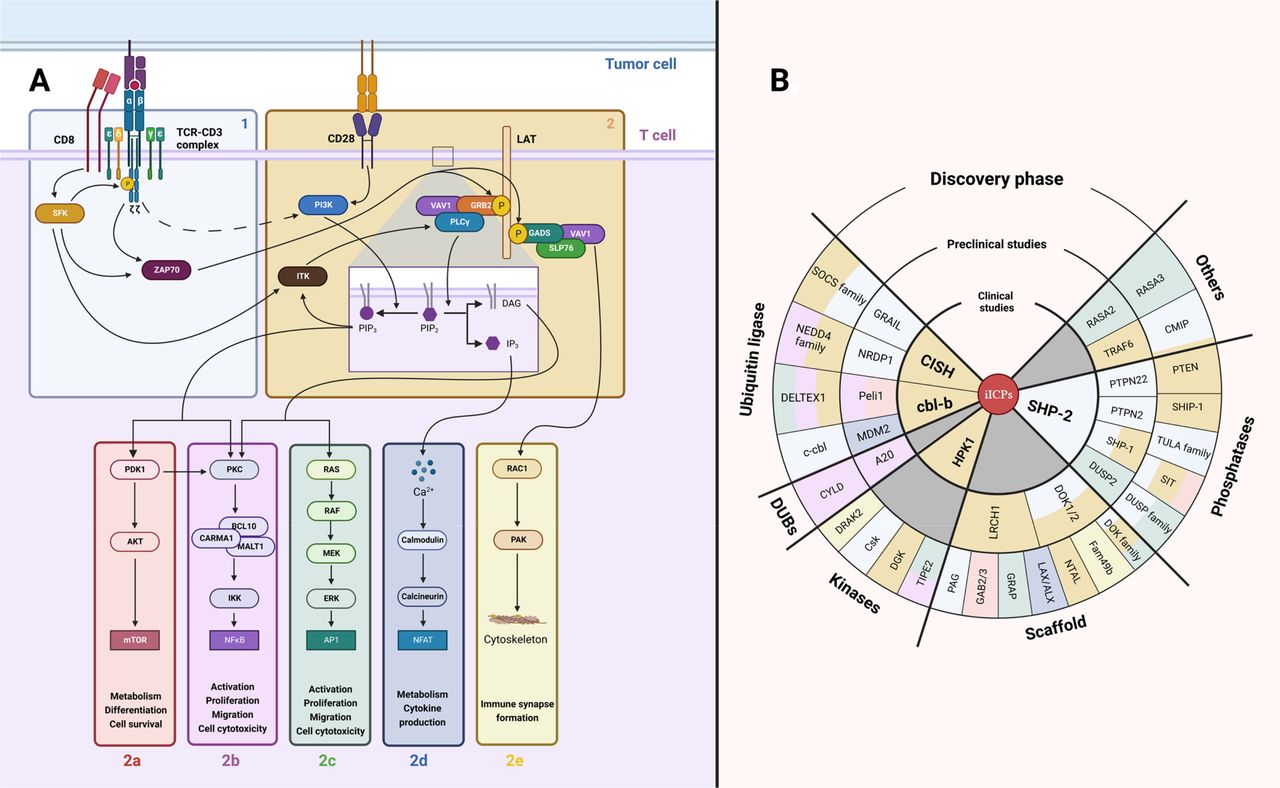 Figure 1
Figure 1 TCR signaling modules. (A) TCR signaling scheme. The first module (1) is responsible for transformation of the interaction of TCR with antigenic peptide associated with MHC class molecule into an intracellular signal. The second module amplifies this signal (2) and further diversifies it (-a–e). Both TCR-CD3 complex (δ, ε, γ, ζ subunits) and CD28 receptor are initially tyrosine-phosphorylated by SFK. (B) TCR signaling inhibiting proteins (iICPs). Numerous negative feedback proteins of TCR signaling were recently discovered. Here, these potential iICPs are classified in order of their clinical approval stage. The color of each iICP backing corresponds to the TCR signaling module where this protein represses the TCR signal. iICP, intracellular immune checkpoint; MHC, major histocompatibility complex; SFK, Src-family protein tyrosine kinases; TCR, T-cell receptor.
The goal of the first step is to transform the interaction of the TCR with antigenic peptide-major histocompatibility complex (pMHC) into an intracellular signal14 (figure 1, box 1). Therefore, this step is responsible for signal initiation. The membrane-embedded TCR/CD3 complex plays a critical role in this process. It consists of TCR α and β subunits which have variable and constant immunoglobulin (Ig)-like domains enabling antigenic peptide recognition. These α and β subunits are non-covalently associated with CD3γ CD3δ and two CD3ε molecules and one signaling domain: an immune receptor tyrosine-based activation motif (ITAM). The CD3ζ homodimer completes the TCR complex with six more ITAMs.15 On TCR engagement, a spatial modification of the TCR complex occurs, allowing the partially phosphorylated Src-family protein tyrosine kinases (SFK) to gain access to ITAMs.16 17 The phosphorylation of ITAMs (pITAM) by SFK family members Lck or Fyn allows the binding of the ZAP-70 SH2-tandem domain.18 ZAP-70 is subsequently phosphorylated by Lck, dissociates from TCR complex and transfers the signal to a second step of the TCR signaling response.19
The aim of the second step is signal amplification and diversification (figure 1, box 2). The key protein at this step is the membrane adaptor LAT. ZAP-70 phosphorylates LAT, leading to the recruitment of numerous adaptor proteins and the formation of the LAT signalosome.20 More than 200 proteins are participating in this signalosome such as SLP76, PLCγ1, VAV1, ITK, RAC1, SOS, PI3K, GRB2 and others.21 Their interactions amplify the initial TCR signal and determine further cell reactivity on TCR engagement. Interestingly, proteins are not the only actors of TCR signaling as phospholipids also play a critical role in T cell activation.22 PI3K phosphorylates phosphatidylinositol 4,5 bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5 trisphosphate (PIP3) which recruits several proteins to the plasma membrane such as ITK that favors PLCγ1 recruitment to LAT signalosome.23 PLCγ1 is responsible for PIP2 hydrolysis into secondary activating molecules inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG).22 At this step, the TCR encoded signal is divided into several major signaling pathways. PIP3 recruits PDK1 to the plasma membrane. This activates the AKT-mTOR pathway responsible for metabolism, differentiation and cell survival (figure 1, box 2a).24 DAG activates PKC and BCL10-CARMA1-MALT1 (CBM) complex that leads to NF-κB nuclear translocation (figure 1, box 2b). In parallel, DAG activates RASGRP that drives RAS-MAPK/ERK pathway (figure 1, box 2c). Both of these pathways are accountable for activation, proliferation, migration and cytotoxicity.25–27 IP3 also binds to its receptor in the endoplasmic reticulum that causes the release of Ca2+ from the endoplasmic reticulum and transcriptional factor NFAT activation (figure 1, box 2d). This latter pathway is responsible for T cell metabolism reorganization and cytokine production machinery.28 Finally, activation of GADS, SLP-76 and VAV1 triggers RAC1 GTPase activation. This allow cytoskeletal reorganization for proper immune synapse formation (figure 1, box 2e).29 Some major contributions of CD28 co-stimulation in TCR signaling networks should be equally noted. Indeed, CD28 can bind directly to PI3K by a well-characterized YMNM binding motif in its cytoplasmic domain30 and will be involved in the AKT-mTOR pathway (box 2a). Moreover, the GRB2/GADS adaptor proteins bind also directly to the CD28 cytoplasmic tail, bridging CD28 to PKC/CBM complex via RLTPR protein.31 This link between CD28 and the GRB2/GADS could also boost some TCR-induced signals (box 2b–e). Beyond this positive signal display, negative feedback mechanisms are set up to establish TCR signal termination.
From negative feedback loops in TCR signaling to bona fide iICPsNegative feedback loops are present to regulate each signaling activation module, thus dampening TCR-induced signal transduction. In the late 1990s, studies on TCR signalosome revealed proteins involved in negative feedback control of TCR signaling, as discussed elsewhere.3 32 The potential to target iICPs emerged after clinical limitations of other cancer immunotherapies such as CAR-T therapies or ICIs, mostly because of intrinsic CD8+ T cell activation suppression due to exposure to numerous immunosuppressive factors of TME (TGFβ, IDO, PGE2, adenosine, ICPs, etc) at the same time.33–35 Targeting these multiple factors by distinct methods could be a complex task. Moreover, these immunosuppressive factors partially act through upregulation of negative signaling protein expression. Therefore, targeting the expression or function of these negative feedback proteins could be a promising method of T cell activation improvement. Preclinical assays on iICPs reviewed below provided encouraging results to improve T cell-based immunotherapies. Targeting of iICPs was facilitated by the rapid progress in cell genetic engineering that occurred this last decade, notably with the availability of CRISPR-Cas9 technology. Thus, a new era is enabled in immunotherapy targeting not only extracellular ICPs but also intracellular molecules—iICPs. This new strategy will complement and can be used in combination with current immunotherapy approaches.
In the following section, we will outline the concept of iICP targeting in cancer immunotherapy. Currently, four of these proteins are targeted in clinical trials and many others are in under preclinical development.
iICPs in clinical trialsDifferent inhibitory feedback loops control the two aforementioned steps of TCR signaling response. Among the proteins reaching clinical trials at phase I, SHP2 targets the initiation step, and three others—CBL-b, CISH, and HPK1—are involved in signal amplification and diversification control.
Modulation of an inhibiting protein may raise safety concerns, particularly with respect to the development of autoimmunity. In the past, before extracellular ICI (CTLA-4, PD-1) blockers came into the clinic, major concerns were raised regarding immune-related adverse events (irAEs).36 These are generally of low intensity, manageable and reversible.37 The example of PD-1 and CTLA-4 and the accumulation of data from preclinical and clinical work will be very beneficial for the future development of iICP targeting. The development of preclinical mouse models and genetic depletion of iICPs in mice certainly helps to appreciate the potential development of autoimmune diseases. This is a crucial step before reaching the clinical steps. However, this is important to keep in mind that PD-1 and CTLA-4 knockout (KO) mouse models have shown some important signs of autoimmune diseases.38–40 Indeed, new retrospective studies involving extracellular ICI show that irAEs may also be associated with a favorable outcome.41 42 Autoimmunity must therefore be carefully considered for each new targeted molecule and in particular iICPs but past experience shows that this does not preclude effective antitumor therapy.
SHP-2Src homology region 2 (SH2) domain containing tyrosine phosphatase-SHP-2 (PTPN11) was shown to be implicated in PD-1-dependent restriction of proximal TCR signaling (figure 2A).43 Hence, similar to ICI antibodies, SHP-2 deletion may relieve TCR signaling inhibition directly at intracellular level. However, SHP-2 deletion was not sufficient to improve clearance of immunogenic tumors even in combination with anti-PD-1 treatment, suggesting an alternative mechanism of PD-1-dependent TCR signal restriction.44 It was demonstrated that another Src homology region 2 domain containing tyrosine phosphatase SHP-1 (vide infra) is also recruited to PD-1 cytoplasmic tail acting in TCR signaling repression.45 Moreover, in the absence of SHP-2, PD-1 recruits SHP-1 to remain functional, suggesting overlapping functions of these proteins.46 Therefore, blocking both SHP-1 and SHP-2 is necessary for TCR signaling improvement. The combination strategy of anti-PD-1 antibody administration with a pharmacological inhibitor of SHP-2 is undergoing clinical trials (figure 2B).
 Figure 2
Figure 2 iICPs in clinical trials. (A) iICPs participating in clinical trials have different action modes. PD-1 engagement activates SHP-2 and leads to repression of proximal signaling events. CBL-B and CISH ubiquitinate their respective targets: regulatory unit of PI3K and PLCγ1, leading to their inactivation. Finally, HPK1 phosphorylates SLP76 that recruits 14-3-3 proteins, following SLP-76 dissociation from LAT signalosome and leading to SLP-76 proteolysis. (B) These iICPs are involved in several clinical trials for cancer treatments. iICP, intracellular immune checkpoint.
CBL-BCbl-b is a member of Casitas B-lymphoma (CBL) family. CBL proteins possess RING finger catalytic domains responsible for protein ubiquitination with sequential degradation of target proteins. CBL proteins lead to the degradation of multiple targets, thus downregulating the TCR signaling cascade.47 48 Cbl-b targets the regulatory subunit p85 of PI3K, interfering with its ability to activate different signaling pathways (figure 2A).49 50 It was shown that Cbl-b represses PTEN inactivation by NEDD4, therefore reducing PI3K activity.51 Cbl-b participates in the regulation of co-stimulatory signal from CD28 or inhibitory receptors CTLA-4 and PD-1.52–55 The loss of Cbl-b on TCR triggering increased Akt/Erk phosphorylation, proliferation, activation, cytokine production (IFNγ, TNFα, IL-2) and cytolytic capacity (Granzyme B).9 54 56–59 TCR-induced proliferation is exacerbated in T cells from children with homogeneous mutations in CBLB gene.60 Cbl-b-deficient mice rejected spontaneous tumor development and adoptive CD8+ T cell transfer from these mice improved control of established or spontaneous tumors from numerous cancer models.9 54 56–59 61 Moreover, both Cbl-b KO CD4+ and CD8+ T cells showed improved resistance to Tregs and TGF-β.57 58 61 Cbl-b was shown to be upregulated in exhausted CD8+ tumor-infiltrated lymphocytes (TILs) and ex vivo abrogation of Cbl-b expression by CRISPR-Cas9 improved cytotoxicity of these cells.9 On in vitro TCR activation, naïve Cbl-b-deficient CD8+ T cells do not require CD28 co-stimulation to be fully activated.57 Finally, CRISPR-Cas9 depletion of Cbl-b in mouse CAR-T cells promotes tumor regression and makes CAR-T cells resistant to exhaustion.9 Therefore, Cbl-b depletion seems to be a potent tool to improve CD8+ T cell–based immunotherapies. Moreover, small molecule inhibitors of Cbl-b activity are under development.62 63 Several clinical trials on Cbl-b inhibition in T cells are ongoing (figure 2B).
CISHA SOCS (Suppressor Of Cytokine Signaling) family protein member negatively regulates CD8+ T cell signaling (figure 2A).64 Indeed, CD8+ T lymphocytes from Cish-deficient mice had improved proliferation, Ca2+ and IL-2) on TCR engagement. These cells had increased expression of effector function associated genes (Il2, Prf1, GrzmB, Eomes, Tbx21, c-Myc and Bcl2l). Moreover, ACT of Cish KO CD8+ T cells enhanced control of tumor progression in tumor-bearing mice.7 Clinical trials targeting CISH by CRISPR-Cas9 in TILs prior to ACT are ongoing (figure 2B).
HPK1Hematopoietic progenitor kinase 1 (HPK1), encoded by the MAP4K1 gene, is a protein kinase identified as a key regulator of TCR signaling. HPK1 is activated by TCR complex on TCR stimulation.65 66 HPK1 associates and phosphorylates SLP-76 at the LAT signalosome. Phosphorylated SLP-76 subsequently binds with GADS and 14-3-3 protein.67 68 This latter association destabilizes the interaction of SLP-76 with LAT signalosome triggering SLP-76 degradation (figure 2A).68 69 This SLP-76 degradation negatively impacts MAPK-ERK pathway signaling.65 70 HPK1 overexpression in a Jurkat T cell line resulted in a MAPK-ERK pathway dampening and suppressing AP-1-dependent gene transcription, notably IL2.65 HPK1 KO and KD in mice resulted in increased T cell proliferation, activation and cytokine secretion, thus granting them the capacity to control tumor growth.71–74 Interestingly, HPK1 expression correlates with T cell exhaustion.10 Furthermore, mouse and human CD8+ and CAR-T cells lacking HPK1 expression showed improved degranulation activity (CD107a), cytokine production and reduced expression of exhaustion markers (PD1, TIM3, LAG3).10 Adoptive cell transfer of mouse and human HPK1 KO CAR-T cells showed improved control of tumor growth in murine xenograft models.10 Ultimately, these data suggest that HPK1 is a crucial regulator of T cell activation of naïve and memory T cells. Numerous small molecule inhibitors of HPK1 are undergoing cancer immunotherapy clinical trials12 (figure 2B).
Targeting iICPs carries a risk of autoimmunity induction. Indeed, aberrant expression of some iICPs led to autoimmunity development in humans.75–77 Invalidation of iICP expression in mice made animals more susceptible to autoimmune disorders.70 78–85 Moreover, ACT of Cish-deficient CD8+ T cells provoke ocular toxicities in mice.7 However, not all iICPs were implicated in autoimmunity development. Notably, CAR-T cells lacking PTPN2 expression showed improved tumor site homing in preclinical models, therefore decreasing the risk of off-target effects and morbidity.86 DRAK2 KO mice showed resistance to autoimmune encephalomyelitis induction despite improved TCR-dependent T cell activation.87 Nevertheless, autoimmunity and toxicity evaluation remain a priority for clinical approval of iICP invalidation for cancer immunotherapy enhancement. After all, such therapies need the development of new strategies of autoimmunity management, allowing reduced toxicities and off-target effects. Besides, tumor-specific targeting improvements might be a key for clinical application of these therapies in the near future.
Although, we present here iICPs as powerful tools to improve antitumor cytotoxic function of T or CAR-T cells, other applications may be envisioned. Notably, their overexpression, potentially, allowing control of CAR-T therapy side effects such as cytokine release syndrome. Indeed, overexpression of Csk (iICP for Lck and Fyn-dependent signal initiation) in TCR-T engineered human T cells undermines TCR signaling and might be used as a safeguard to prevent excessive activation of immune cells during ACT.88
Other TCR iICPs with completed preclinical trialsRecently, other TCR iICPs showed promising results and improved T-cell-based immunotherapies in animal tumor models bringing their targeting close to clinic. Below are listed proteins involved in negative signals downstream TCR triggering and where mouse models are used to highlight the potential iICP status of these molecules. Due to their mechanism of action, these proteins could be divided into different groups: lipid kinases, protein phosphatases, ubiquitin ligases, deubiquitination enzymes (DUBs), hydrolases and scaffold proteins.
Lipid kinasesDGKα and ζ (diacylglycerol kinases)DGKs are enzymes that phosphorylate DAG, a second messenger molecule in TCR signaling generated by activated PLCγ1.89 This interrupts DAG association to RasGRPs, inducing RAS/MAPK pathway blockade and attenuates TCR signaling.90–92 DGKζ KO CD8+ mice had significantly improved resistance to tumor growth, associated with increased CD8+ T cell infiltration. Moreover, mice receiving an ACT of naïve or primed DGKζ CD8+ T cells exhibited improved in vivo antitumor responses.90 93 DGK KO or pharmacological inhibition improved CAR-T cell cytotoxicity against tumors both in mouse models and in human CAR-T cells.8 93 94 CRISPR inactivation of two isoforms of DGK in CAR-T cells synergistically improved in vivo tumor clearance, cytokine production, proliferation and reprogrammed CAR-T cells to effector memory phenotype.8
Protein phosphatasesNon-receptor protein tyrosine phosphatases (PTPN)Among this large PTPN family, at least three members could be considered involved in negative feedback loops downstream TCR signaling and were previously tested in the context of anticancer activity in preclinical models: PTPN2, PTPN6 and PTPN22. PTPN2, also known as TC-PTP, is a PTP mainly expressed in hematopoietic cells and involved in T cell signaling.95 PTPN2 directly dephosphorylates Lck and Fyn (SFK members) kinases both in CD4+ and CD8+ T cells establishing a threshold for TCR triggering.96 PTPN2 deletion in mouse T cells prevents tumor formation in a p53+/- mouse model.86 PTPN2 KO T cells had enhanced T cell–mediated immunosurveillance, increased effector memory T cell numbers, tumor infiltration and produced more cytokines.86 PTPN2 deletion in mouse CAR-T cells lead to effector memory phenotype (CD44+CD62LNEG) and increased expression of IFNγ, TNFα and Granzyme B making them less prone to exhaustion. CAR-T cells lacking PTPN2 were more efficient in eradicating solid tumors in mice.86 Moreover, small molecule inhibitor of PTPN2 improved mouse CAR-T cell cytotoxicity as well as it was the case for human CAR-T cells in vitro.86 97 PTPN6 is also well known as SHP-1 (Src homology 2 domain-containing protein tyrosine phosphatase 1). The specific deletion of the phosphatase SHP-1 in naïve CD8+ T cells enhances their proliferation potential, cytolysis capacity in vivo and improved IFNγ, TNFα, IL-2 production.98 99 ACT of these cells augmented mice survival in disseminated FBL leukemia model.100 However, no difference was found in tumor progression in solid tumor model of melanoma. Intriguingly, implementation of PD-1 blockade demonstrated that SHP-1 KO CD8+ T cells were more responsive to anti-PD-1 and had improved control of melanoma B16-F10 cell growth.101 Moreover, SHP-1 (and partially SHP-2) pharmacological inhibition improved cytotoxic capacity of human primary CD8+ T cells against tumor.102 As SHP-1 is the closest homolog of SHP-2 and can play some similar functions in T cells,46 it would be of interest to test a potent dual SHP-1/-2 inhibitor in these preclinical models. PTPN22 can also dephosphorylate SFK members at their activation sites inhibiting TCR signaling initiation.103 104 In ACT experiments, it was demonstrated that primed CD8+ PTPN22 KO mouse T cells controlled better tumor growth, produced more cytokines and were highly resistant to immunosuppressive effects of TGFβ.105–107
DUSP2 (dual specificity phosphatase 2)DUSP family member DUSP2 (PAC1) is upregulated in exhausted tumor-infiltrated T lymphocytes. Indeed, DUSP2 KO mouse CD8+ TILs showed less exhaustion markers (PD-1, TIM-3, LAG3), improved IFNγ, TNFα, Granzyme B production, tumor growth control and had enhanced survival.108 Other DUSP family members (such as DUSP14, DUSP22) could be also involved in the inhibitory feedback control of the signals encoded by the TCR triggering. However, their iICP capacities are not documented by mouse tumor models.
Ubiquitin ligasesMDM2 (murine double minute 2)MDM2 is E3 ubiquitin ligase responsible for degradation of NFATc2. Naïve CD4+ T cells from MDM2 KO mice showed enhanced IL-2 and IFNγ production on TCR stimulation. Adoptive CD4+ T cell transfer decreased tumor growth in tumor-bearing mice.109
NRDP1NRDP1 takes part in ZAP-70 ubiquitination.110 This promotes the recruitment of STS1 and STS2 phosphatases, which leads to ZAP-70 dephosphorylation. On TCR stimulation, CD8+ T cells from Nrdp1-deficient mice had improved proliferation, increased signaling protein phosphorylation (ZAP-70, LAT, PKC, ERK-1/2 and JNK-1/2), cytokine production (IFNγ, IL-2), higher expression of key transcriptional factors Prf1 (perforin), Gzmb (Granzyme B), T-bet, Eomes, associated with effector function of CD8+ T lymphocytes. Moreover, Nrdp1-deficient primed CD8+ T cells had a better control of syngeneic tumor development in a mouse model during adoptive cell transfer.110
GRAIL (gene related to anergy in lymphocytes)Ubiquitin ligase GRAIL directly targets the TCR complex leading to TCRβ and CD3ζ subunit degradation. Mouse Grail-deficient CD4+ T cells had increased proliferation, activation, survival and resistance to anergy induction on TCR activation.111 GRAIL KO mice better controlled tumor growth in an experimental model due to improved CD8+ T lymphocytes cytotoxic activity. Notably, CD8+ TILs lacking GRAIL had improved IFNγ and Granzyme B production and increased expression of IL-21R.112
Peli1 (Pellino E3 ubiquitin protein ligase 1)E3 ubiquitin ligase Peli1 negatively controls TCR signaling by two distinct ways. (1) On TCR stimulation, it targets c-Rel protein of NFκB family responsible for T cell activation, proliferation and cytokine production by ubiquitination leading to degradation.79 (2) After TCR engagement, Peli1 mediates ubiquitination of TSC1 that improves TSC1/TSC2 dimerization. TSC1/TSC2 dimerization inhibits mTORC1, a protein of PI3K-Akt pathways known for its metabolic regulations.113 Recently, it was shown that Peli1 KO mice better control tumor growth in different tumor models due to higher CD4+ and CD8+ T cell tumor infiltration and enhanced cytokine production (IFNγ, granzymes) in these cells.113
Deubiquitination enzymesA20The ubiquitin-editing enzyme A20 (also known as tumor necrosis factor-α-induced gene 3, TNFAIP3) removes ubiquitin chains on activated MALT1 in the CBM complex (see box 2b in figure 1). Deubiquitinated MALT1 does not interact with IKK stopping NFκB activation on TCR stimulation.114 A20 KO CD8+ T cells demonstrate higher cytokine production (IFNγ, TNFα, IL-2) and cytotoxicity (Granzyme B). ACT of in vitro pre-stimulated A20 KO CD8+ T cells shows a significant reduction of tumor growth in mouse melanoma model.115 116
HydrolasesRASA2 (RAS p21 protein activator 2)Genome wide CRISPR screen in primary human CD8+ T cells reveals that Ras-GTPase RASA2 KO enhances human CD8+ T cell proliferation and in vitro anti-cancer function.117 Recently, RASA2 ablation improved in vivo tumor control during adoptive cell transfer of engineered T cells in multiple xenograft models.118
Scaffold proteinsDok (Downstream of kinases) familyMembers of Dok (for Downstream of kinases) family proteins play a role in negative regulation of TCR signaling.3 119 120 For instance, Dok1 and Dok2 proteins recruit different negative enzymes such as Csk, SHIP-1 or Ras-GAP establishing a platform for these proteins and recruiting them in close proximity to the LAT signalosome. Recently, our group demonstrated that Dok-1/2 exert their negative role mainly in primed CD8+ T cell showing an improvement of Akt and Erk phosphorylation on TCR engagement. Unexpectedly, Dok-1/2 KO mice did not improve tumor cell cytotoxicity in vitro and in vivo, probably due to re-wiring of T cells signaling in absence of Dok-1/2.121
LRCH1 (leucine-rich repeats and calponin homology domain containing 1)LRCH1 is a negative regulator of TCR signaling that binds directly to LAT, disturbing LAT signalosome leading to LAT endocytosis. LRCH1 deficiency improves TCR signaling in CD8+ T cells. CD8+ T cells lacking LRCH1 have increased cytokine production, activation and proliferation on TCR stimulation. ACT of LRCH1 KO CD8+ T cells improved tumor control in mice. LRCH1 invalidation by CRISPR-Cas9 in human primary T cells improved IFNγ production, proliferation and migration of these cells.122
Unknown mechanismTNF receptor-associated factor 6TRAF6 is an adaptor protein that mediates numerous protein-protein interactions and a RING E3 ubiquitin ligase. TRAF6 negatively regulates PI3K signaling.84 On the contrary, TRAF6 is important in CBM (Carma1-Bcl10-MALT1) complex formation necessary for IKK activation and nuclear translocation of NFκB.123 Lack of NFκB results in impaired maturation and activation of regulatory T cells (Treg), known for their immunosuppression in tumor sites.124 Treatment of tumor-bearing mice with TRAF6 interaction peptide inhibitor improved cytokine production in TILs, restrained tumor development in mice, that was associated with restricted Treg migration into tumors.125
Other perspective for clinical application of iICPsWe review here more than 30 TCR signaling inhibitory intracellular proteins. The vast majority of them were discovered in the last 5 years and remain under intense investigations prior to validation in preclinical tumor models. The summary of these proteins could be found in table 1. The mouse tumor models used here highlight the possibility to target genetically the potential iICP gene expression in TILs or CAR-T cells for developing clinical trials (see discussion below). However, a pharmacological iICP inhibition could also be taken into account. Some syngeneic mouse models could be used to evaluate a broad impact of these inhibitors, as it was shown for MDM2 inhibitors which promoted the recognition of tumor cells by T cells.126 127
Table 1Other perspective for clinical application of iICPs
DiscussionThe development of gene engineering and synthetic biology extend greatly the possibilities of cancer immunotherapies. Indeed, immunotherapies are based on the fundamental immunology knowledge and technical possibilities. Gene modification tools such as TALEN or CRISPR-Cas9 and their validation in clinical trials extended our clinical interest beyond the cell surface. Genetically modified cells have been approved recently for clinical use. Consecutively, it opened new avenues to engineer more extensively T cells, reaching previously inaccessible targets such as iICPs. Currently, numerous tools to modulate iICPs expression or activity are available (figure 3): small molecules able to inhibit the activity of iICPs, methods of in vivo targeted proteolysis of iICPs (PROTAC), in vitro gene silencing by interference (siRNA, ASO, CRISPR interference) and gene editing (CRISPR-Cas9, TALEN). These methods could be realized in different vector and non-vector-based delivery approaches. These technologies are constantly improving, thus expanding the toolbox to develop new strategies for immunotherapies.
 Figure 3
Figure 3 iICP targeting. Numerous methods to target iICPs were developed recently for in vitro/ex vivo and in vivo application in line with their possible clinical use: adoptive cell transfer (TILs, CAR-T, TCR-T) and systemic therapy. Targeting could be performed at different levels of iICP protein expression or activity. It could be done by irreversible gene modification or temporary inhibition. It could act on iICP expression, protein function or key interactions with partner proteins. CAR-T, chimeric antigen receptor T cell; iICP, intracellular immune checkpoint; TCR, T cell receptor; TIL, tumor-infiltrated lymphocyte.
Modern T cell–based immunotherapeutic approaches use different tumor-specific antigen receptors such as conventional TCRs (TILs), engineered TCRs (TCR-T), chimeric antigen receptor (CAR) or brand new T cell receptor fusion constructs (TRuC), that combine TCR and CAR by expression of one of TCR chains fused to scFv fragment.128–132 However, each antigen receptor type differs from others in antigen sensitivity, triggering mechanisms, immune synapse formation and signaling pathways. All these mechanisms need further investigations as adequate understanding of antigenic receptor signaling in each case could improve clinical outcome in patients. Recently, several data compared the first clinically validated artificial antigenic receptor, CAR, with conventional TCR signaling.11 133 Actually, CAR and TCR use similar signal transduction molecular pathways but the magnitude and kinetics of phosphorylation events are different.134 135 In this context, it is also important to know if these iICPs are able to control the encoding signals downstream of CAR. Some of these proteins, involved in negative feedback loops downstream the TCR signaling, have been challenged in different kinds of CARs. Indeed, targeting PTPN2, DGK, HPK1, and Cbl-b is also efficient in the context of CAR-T cells.8–10 86 Moreover, CAR structure might impact signalosome formation as 4-1BB-CAR recruit Themis-SHP1 complex, but it is not the case for CD28-CAR.136 On the contrary, PD-1-SHP2 has greater suppressor effect in CD28-CAR.137 Therefore, the construction of T cell antigenic receptor is crucial for signaling and subsequent biological effect, and this should be considered for improvement of T cell activation via targeting of TCR signaling inhibitory loops.
Most of the early studies on T cell signaling were performed on T cell lines and primary CD4+ T cells.3 However, the role of the iICPs should be considered in different T cell subsets involved in immunotherapy such as exhausted CD8+ T cells inside the tumor or in vitro expanded T cells in the context of CAR-T cells and ACT. Some differences in TCR signalosome formation could be suggested among T cell subsets. The first evidence of TCR signalosome difference between naïve and memory T cells was revealed as memory CD4+ T cells had less tyrosine phosphorylated proteins on TCR engagement, notably ZAP-70.138 These data suggest that there is a rearrangement of TCR signalosome on the passage to the memory state, meaning that inhibitory mechanisms of TCR signal propagation may have specificity toward the naïve or memory TCR configuration. However, the composition and dynamics of the proximal TCR signal transduction protein network seems to be largely conserved between human expanded CD4+ and CD8+ T cells.139 And for instance, the iICP, HPK1 binds to SLP-76 on TCR triggering with similar kinetics in human expanded CD4+ and CD8+ T cells.139
ACT therapy for cancer treatment is rapidly expanding notably after the clinical acceptance of CAR-T cells. Other immune cells with cytotoxic potential are currently being tested. This is the case for NK and γδ T cells.140–143 Recently, the engineering and clinical efficacy of CAR-NK or CAR- γδ T cells were demonstrated.144–147 Interestingly, it is known that NK and γδ T cells activating receptors (NCRs, NKG2D and γδ TCR) share similar signaling activating machinery as αβ TCR.148 149 Therefore, several signaling inhibiting loops are shared and impact the activation of these cells. Targeting these inhibiting proteins may improve ACT using NK and γδ T cells. Notably, Cbl-b-deficient NK cells show improved cytotoxicity, antitumor immunity and metastasis control in vivo.150 NK cells lacking HPK1 have increased cytotoxicity against NK sensitive murine lymphoma cell, YAC-1.72 DOK1 and DOK2 are induced on NK activating receptor engagement and their ablation enhanced IFNγ production after stimulation.151 Recently, our team showed that CISH depletion specifically in NK cells improves NCR signaling, proliferation, cytokine production and antitumor activity in vitro and in vivo.152 Furthermore, the investigation of other inhibitory proteins implications in NK and γδ T cell activation may open a large window of opportunities for ACT cancer immunotherapy improvements.
In this review, we were focused on iICPs that negatively control TCR signaling. However, T cell activation is a complex process including cytokine signaling as well. It was demonstrated that some iICPs such as PTPN2 and CISH might negatively regulate cytokine signaling pathway JAK-STAT, therefore contributing to T cell activation improvement also by this mechanism.153 154
ConclusionImmunotherapies are showing encouraging results in disease management for patients with cancer. Historically, most efforts are focused on targeting molecules expressed at the surface of immune cells. As described here, there are several promising avenues to target intracellular molecules. We have now entered the era where cell therapy and genetic modification of a cell is possible. These approaches will complement current immunotherapy strategies and can also be used in combination with other treatments such as those based on immune checkpoint inhibitor, CAR-T cells, targeted therapies and more. However, immune cell signaling needs to be studied in detail prior to propose these new innovative treatments.
Although we focused on targeting iICPs in T cells in this review, other cytotoxic cells may be used in the future. NK and γδ T cells have different properties in tumor cell recognition, alloreactivity or persistence. Interestingly, these cytotoxic lymphocytes share some mechanisms of signaling and activation with conventional T cells, thus targeting aforementioned iICPs may be of interest in these cells. Inhibitory proteins mentioned in this review and proteins not yet identified or studied in the context of TCR signaling have a great potential to improve existing cell-based immunotherapies of cancer and are expected to upgrade cancer treatments in the near future.
Ethics statementsPatient consent for publicationEthics approvalNot applicable.
AcknowledgmentsThe authors wish to thank Thomas Miller (CRCM, Marseille) for critical reading and to improve language quality of the manuscript.
留言 (0)