
記住我
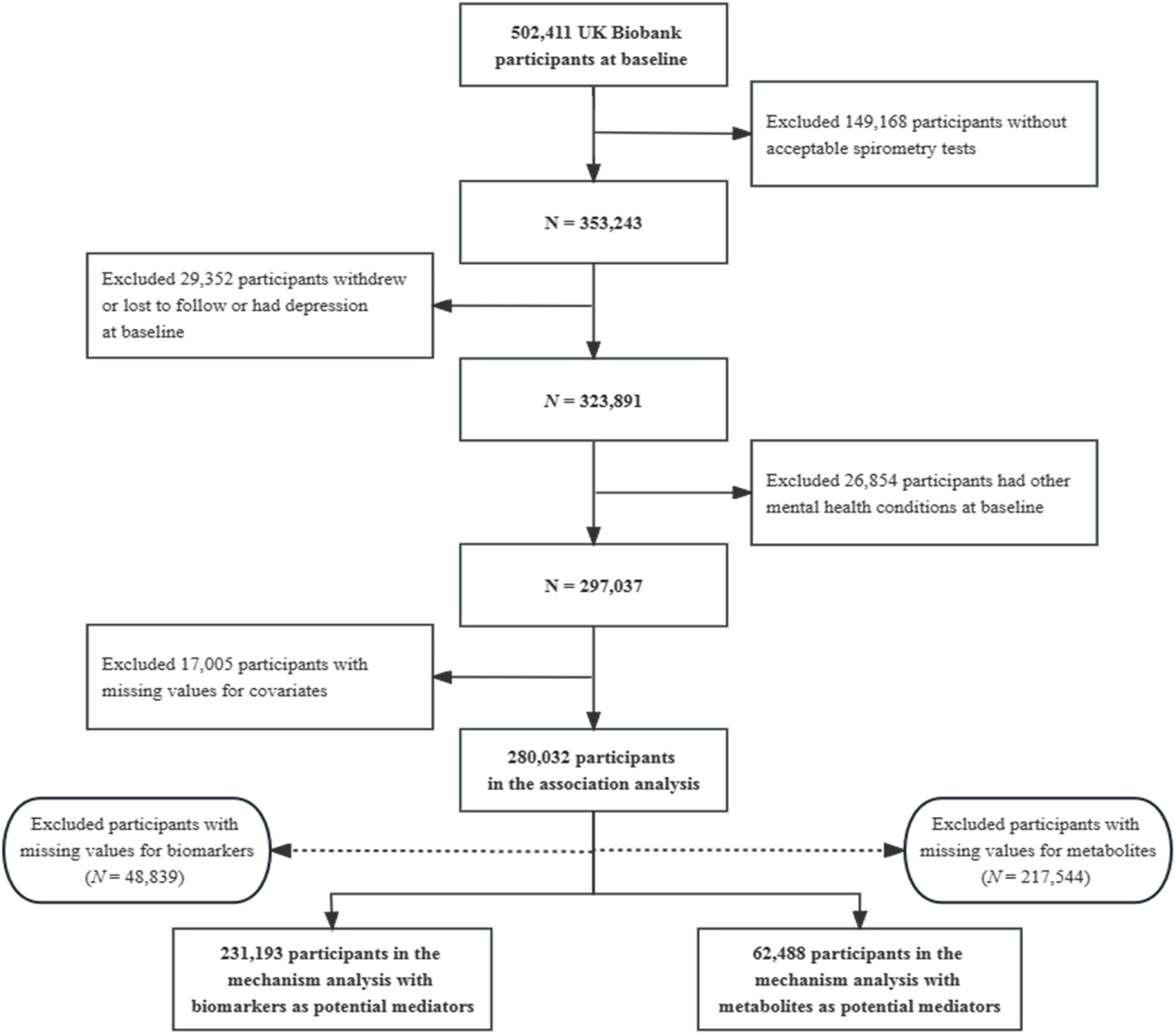


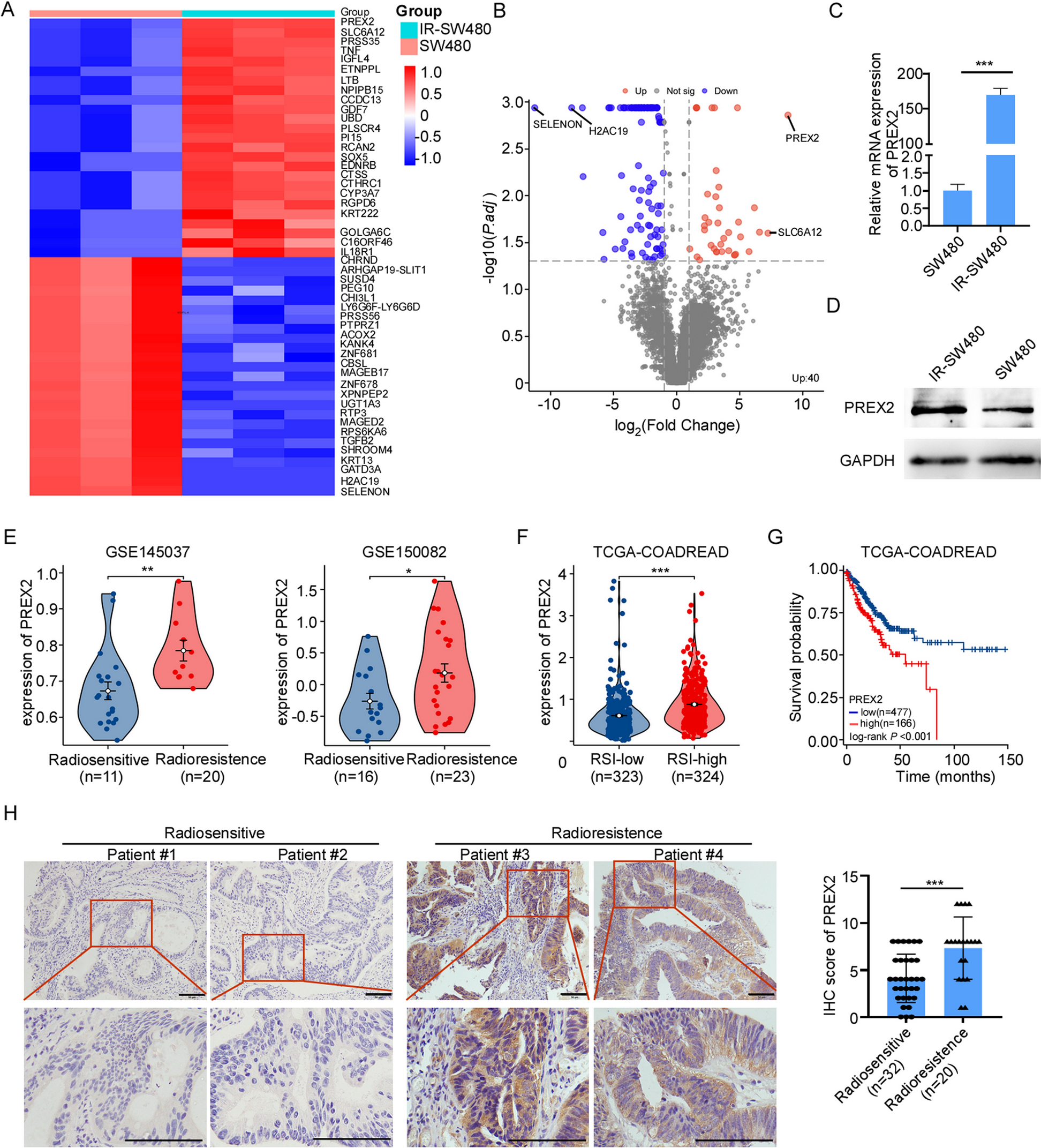
First, we evaluated the quality of the data, and performed a PCA. The data in GSE57691 exhibit satisfactory repeatability (Fig. 1A). In order to screen for differentially expressed autophagy-related genes, we next analyzed the expression of 232 autophagy-related genes in GSE57691, and 20 autophagy-related genes were identified using the criteria of adjusted P-value < 0.05 and |log2FC|≥ 1, including 1 up-regulated genes and 19 down-regulated genes, which were presented in heatmap and volcano plot (Fig. 1B and C).
Fig. 1
Differentially expressed autophagy-related genes in PAD and healthy samples. A Principal component analysis for GSE57691. B Heatmap of the 20 differentially expressed autophagy-related genes in PAD and healthy samples. C Volcano plot of the 20 differentially expressed autophagy-related genes. The red dots represent the significantly up-regulated genes and the blue dots indicate the significantly down-regulated genes
PPI network and correlation analysis of the differentially expressed autophagy-related genesTo determine the interactions among differentially expressed autophagy-related genes, we performed PPI analysis. The proteins corresponding to the genes interact biochemically with each other in at least one and up to 10 different ways (Fig. 2A). To explore the expression correlation of these autophagy-related genes, Spearman correlation analysis was performed, yielding a potential interaction of 20 differentially expressed autophagy-related genes in the GSE57691 dataset (Fig. 2B).
Fig. 2
PPI, Spearman correlation analysis, and validation of the 20 differentially expressed autophagy-related genes. A The PPI among 20 differentially expressed autophagy-related genes. B Spearman correlation analysis of the 20 differentially expressed autophagy-related genes. C Histogram of mRNA expression levels of the autophagy-related genes in WalkByLab participates. Samples with Ct values of reference genes greater than 25 were removed. *P < 0.05; **P < 0.01; ***p < 0.001; ns, non-significant
Validation of differentially expressed autophagy-related genes in WalkByLab registry PAD patientsThe expression levels of these differentially expressed autophagy-related genes were further validated by qRT-PCR analyses of PBMCs from our cohort of WalkByLab Brandenburg (PAD, n = 18; non-PAD controls, n = 18. Clinical characteristics available in Additional file 1: Table S2). Expression levels of BNIP3, EEF2, FOXO1, HSPA5, KLHL24, MAP1LC3B, PEA15, PTEN, RAB11A, RB1CC1, RPS6KB1, SAR1A, ST13, and VAMP3 genes were significantly lower in PAD patients as compared to non-PAD controls (Fig. 2C). The BAG3, CCL2, CSTB, and EEF2K genes tended to be less expressed in the PAD group but did not reach statistical significance. However, Expression levels of CX3CL1 and HSPB8 genes were very low, so we did not include them in the statistical analysis.
GO and KEGG enrichment analysis of the differentially expressed autophagy-related genesTo analyze the potential biological functions of these differentially expressed autophagy-related genes, we conducted GO and KEGG enrichment analysis. The results revealed that the most significant GO enriched terms are linked to the biological functions as follows: 1) response to nutrient levels, process utilizing autophagic mechanism, autophagy (biological process); 2) transport vesicle, aggresome, chaperone complex (cellular component); and 3) cell adhesion molecule binding, GTPase activity, ubiquitin-like protein ligase binding (molecular function) (Fig. 3A; Additional file 1: Table S3). In KEGG enrichment analysis, the differentially expressed autophagy-related genes are mainly involved in the process of autophagy and AMPK signaling pathway (Fig. 3B and C; Additional file 1: Table S4).
Fig. 3
GO enrichment and KEGG analysis of 20 differentially expressed autophagy-related genes. A GO enrichment analysis. B and C KEGG analysis
Autophagy levels of PBMC in WalkByLab registry PAD patientsSo far, our analysis revealed that autophagy-related genes’ mRNAs are expressed at low levels in PBMCs of PAD patients and their potential biological functions are mainly associated with autophagy according to GO and KEGG analysis. Next, we assessed the expression of important autophagy markers (p62, beclin1, and LC3B) to explore the autophagy level between PAD and health.
First, the mRNA levels of p62, beclin1, and LC3B were examined in GSE57691 No significant differences in the mRNA levels of p62 and beclin1 were detected between PAD patients and healthy controls. However, the gene expression level of LC3B was found to be significantly decreased in PAD patients (Fig. 4A). Next, we validated a possible deregulation of LC3B mRNA expression in PBMCs from WalkByLab patients using qRT-PCR. LC3B in WalkByLab participates was less expressed in the PBMCs of patients with PAD (Fig. 4B). Finally, we assessed the protein levels of p62, beclin1 and LC3B in the PBMCs of WalkByLab participates. It turns out that there was a significant decrease in beclin1 and LC3B-II protein levels in PAD patients as compared to healthy controls (Fig. 4C). There was a trend of increased p62 level, but it was not statistically significant. In summary, results showed that protein levels of autophagy markers were decreased in PBMCs from patients with PAD.
Fig. 4
Autophagy levels in PAD patients and healthy controls (HC). A mRNA levels of autophagy markers (p62, Beclin1, and LC3B) in GSE57691. B qRT-PCR analysis of mRNA levels of autophagy marker LC3B in WalkByLab participates. C Western blotting analysis of protein levels of autophagy markers (p62, Beclin1, and LC3B) in WalkByLab participates. *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant
Quantitative analysis of immune microenvironmentPAD is a systemic disease of the arteries, which manifests in the periphery (e.g. legs, feet) but usually affects the majority of all body arteries. PAD is also an inflammatory disease, and the degree of systemic inflammation is even greater than in CAD due to the large number of arteries affected [17]. Thus, we performed a ssGSEA here to quantify the immune microenvironment of the artery wall and compared tissue from PAD patients with healthy controls. ssGSEA revealed 16 types of immune cells and 13 types of immune-related pathways in each sample, which is shown in the heat map (Fig. 5A). A higher level of immune cell infiltration (including aDCs, iDCs, Mast cells, neutrophils, NK cells, pDCs, T helper cells, Tfh, Th1, and Th2 cells) was identified in PAD as compared with healthy controls (Fig. 5B). Moreover, more active immune-related pathways (including APC co-stimulation, cytokine-cytokine receptor (CCR) interaction, check point, MHC class I, and T cell co-stimulation activity) were observed in PAD as compared with healthy controls (Fig. 5C). Further Spearman’s correlation analysis showed that the identified subset of infiltrating immune cell subgroups and active immune-related pathways were moderately to strongly correlated, respectively (Fig. 5D and E).
Fig. 5
The landscape of immune microenvironment between PAD patients and healthy controls based on ssGSEA. A A heatmap of 16 immune infiltration cells and 13 immune-related pathways. B Boxplot of comparisons of 16 immune infiltration cell enrichment scores. C Boxplot of comparisons of 13 immune-related pathways enrichment scores. D Correlation matrix of ssGSEA scores of immune cells. E Correlation matrix of ssGSEA scores of immune-related pathways. F Correlation analysis between the expression of 20 differentially expressed autophagy-related genes and immune microenvironment. Red means positive correlation and blue means negative correlation. *p < 0.05, **p < 0.01, ***p < 0.001, ns, non-significant
Next, we analyzed whether these 20 differentially expressed autophagy-related genes were correlated with immune function in PAD. We found that most autophagy-related genes (except for KLHL24, HSPB8, CX3CL1 and CCL2) correlated with immune cells infiltration and immune-related pathways (Fig. 5F). Among them, RB1CC1 and FOXO1 genes showed the highest reported number of relationships with immune microenvironment. Most infiltrating immune cells linked to autophagy-related genes were PBMCs such as iDCs (involved genes n = 6), pDCs (n = 8) and Th2 cells (n = 9). The top immune-related pathways associated with autophagy-related genes were found to be CCR interactions (n = 10), as demonstrated in Fig. 5F. Specifically, the following genes were identified as being involved in these pathways: VAMP3, ST13, RPS6KB1, RB1CC1, PEA15, MAP1LC3B, HSPA5, FOXO1, EEF2K, and BNIP3.
Expression level of chemokines in plasmaSince CCR interaction was the top active immune-related pathway that linked to autophagy-related genes in the ssGSEA analysis, and combined with the results that autophagy-related genes and autophagy level were decreased in PAD patients’ PBMC, we suspect that CCR interaction might play an important role in the development and progression of PAD. Among CCR interactions, chemokines are best known for their ability to stimulate the migration of leukocytes. Since leukocytes are central to the progression of PAD and to our analysis of autophagy in PBMCs, chemokines play a particularly central role in our investigation.
Hence, we used the human chemokine antibody array to explore the plasma levels of chemokines (3 PAD and 3 health controls) and found that growth-related oncogene (GRO) and neutrophil activating protein2 (NAP2) chemokines had highest fold change expression between the two groups (Fig. 6A and B). The mRNA levels of GRO family (GROα, GROβ and GROγ) and NAP2 were also highly expressed in PAD patients in GSE57691 (Fig. 6C). Thus, we next utilized ELISA kits to measure the plasma expression levels of GRO and NAP2 in WalkByLab registry cohort of 179 participants. GRO and NAP2 levels were significantly higher in PAD patients’ plasma, and NAP2 (AUC: 0.752) demonstrated a better diagnostic performance for PAD patients as compared with GRO (AUC: 0.670) (Fig. 6D). Similar results were also shown in the propensity score matching analysis cohort after excluding the potential influence of age (Fig. 6E).
Fig. 6
The plasma levels of chemokines in PAD and control groups and the relationship between autophagy-related genes, chemokines and endothelial function. A The expression profiles of human chemokines in PAD and health controls’ plasma in WalkByLab participates. Chemokines are ranked according to the value of the log2-fold changes between the two groups. B The relative protein expression of GRO and NAP2 in PAD and health controls. C The mRNA expression levels of the three subtypes of GRO and NAP2 in GSE57691. D The plasma levels of GRO and NAP2 as well as their diagnostic values for PAD in the whole cohort of 179 participants. E The plasma levels of GRO and NAP2 in the PSM cohort of 68 participants. F–H Spearman correlation analysis between autophagy-related genes and chemokines and FMD. I Spearman correlation analysis between chemokines and FMD. *P < 0.05; **P < 0.01; ***p < 0.001; ns, non-significant
The relationship between autophagy-related genes, chemokines and endothelial functionSpearman correlation analysis, which investigate the potential pairwise correlation between these three factors, showed that autophagy-related genes KLHL24, VAMP3, HSPA5, and ST13 were moderately negative correlated with chemokine GRO (|Cor|> 0.4) (Fig. 6F), and showed mild to moderate negative correlation with chemokine NAP2 (|Cor|> 0.3) (Fig. 6G). Fourteen autophagy-related genes were moderately positive correlated with FMD (Cor > 0.4) (Fig. 6H). However, the chemokines GRO and NAP2 were not correlated with FMD (P > 0.05) (Fig. 6I).
The correlation between plasma chemokines and walking capacityGardner treadmill testing is a gold standard exercise stress test, providing clinically important diagnostic and prognostic information for patients with symptomatic PAD [18]. The results of the Spearman correlation analysis revealed that there were significant correlations between the levels of GRO and NAP2 and the walking capacity. Specifically, the GRO level showed a mild negative correlation with pain-free walking distance (Correlation coefficient, Cor = -0.331) and maximum walking distance (Cor = -0.340). The NAP2 level demonstrated a mild to moderate negative correlation with pain-free walking distance (Cor = -0.387) and maximum walking distance (Cor = -0.403). Besides, there was a moderate positive correlation between the levels of GRO and NAP2 (Cor = 0.434), and a strong positive correlation between pain-free walking distance and maximum walking distance (Cor = 0.913). The correlation network plots for pairwise correlations among the studied variables are showed in Fig. 7A (all P < 0.05).
Fig. 7
The relationship between chemokines, autophagy-related genes, and treadmill walking capacity. A The correlation network plots for pairwise correlations among GRO, NAP2 and walking distance in Gardner treadmill testing. B Participates with walking time less than 6 min in Gardner treadmill testing had higher levels of GRO and NAP2 protein. C The predict values of GRO and NAP2 for impaired walking capacity in Gardner treadmill testing. D Spearman correlation analysis between autophagy-related genes and walking distance in Gardner treadmill testing. *P < 0.05; **P < 0.01; ***p < 0.001; ns, non-significant
Furthermore, participates which showed during the walking test a walking time less than 6 min had higher levels of GRO and NAP2 protein (Fig. 7B). ROC analysis to determine the diagnostic performance in walking time less than 6 min showed that NAP2 (AUC: 0.743) had a higher efficacy than GRO (AUC:0.701) (Fig. 7C).
The correlation between autophagy-related genes and walking capacitySpearman correlation analysis showed that, except for CCL2 and CSTB genes, all other autophagy-related genes exhibited a significant positive correlation with walking distance, with correlation coefficients ranging from moderate to strong (0.40 < Cor < 0.73) (Fig. 7D). Specifically, MAP1LC3B gene demonstrated strong positive correlations with pain-free (Cor = 0.70) and maximum walking distance (Cor = 0.73).
The correlation between plasma chemokines and major adverse cardiovascular events (MACE)We reviewed the history of MACE occurrence, including stroke, transient ischemic attack, and myocardial infarction, among the enrolled participants. Our results showed that there was no statistical difference in NAP2, GRO, and ankle-brachial index (ABI) levels between the groups with and without the history of MACE occurrence (P > 0.5) (Additional file 2: Figure S1). The AUC of GRO was 0.529 and that of NAP2 was 0.516, which is marginally higher than that of ABI (AUC: 0.508). Overall, these biomarkers almost had little predictive power for MACE.
Construction of a nomogram to predict impaired exercise toleranceNomogram is a powerful tool used for predicting the incidence of diseases or patients’ prognosis. Here, we constructed a nomogram model to predict impaired walking capacity. A walking time < 6 min in Gardner treadmill testing was defined as the endpoint. LASSO regression was performed firstly to select the most effective variables from all clinical features and ten clinical parameters were obtained (Fig. 8A). The respective AUC values of these parameters were shown in Fig. 8B.
Fig. 8
Construction of nomogram model. A LASSO regression analysis selected the potential variables. B ROC curve shows the predicted ability of the chosen variables. C Nomogram integrated with NAP2 predicting walking time < 6 min in Gardner treadmill testing. D ROC curve analysis compares the predicted efficacy of different nomogram models. E Calibration curves for nomogram predicted walking time < 6 min in Gardner treadmill testing
Given the significant correlation between the levels of GRO and NAP2 and walking capacity, we selected these two factors as candidate variables for multivariate logistic regression analysis and nomogram construction (Fig. 8C and Additional file 2: Figure S2). Due to the limited sample size available for the autophagy-related genes in our study, we opted not to include them as candidate variables for analysis. ROC curves analysis showed that the nomogram integrated with NAP2 (AUC: 0.860) had a better predicted efficacy than nomogram with GRO (AUC: 0.854) (Fig. 8D). Finally, calibration plots were used to visualize the performances of the nomograms. The calibration plot confirmed the performance of our predictive model (Fig. 8E).
留言 (0)