
記住我
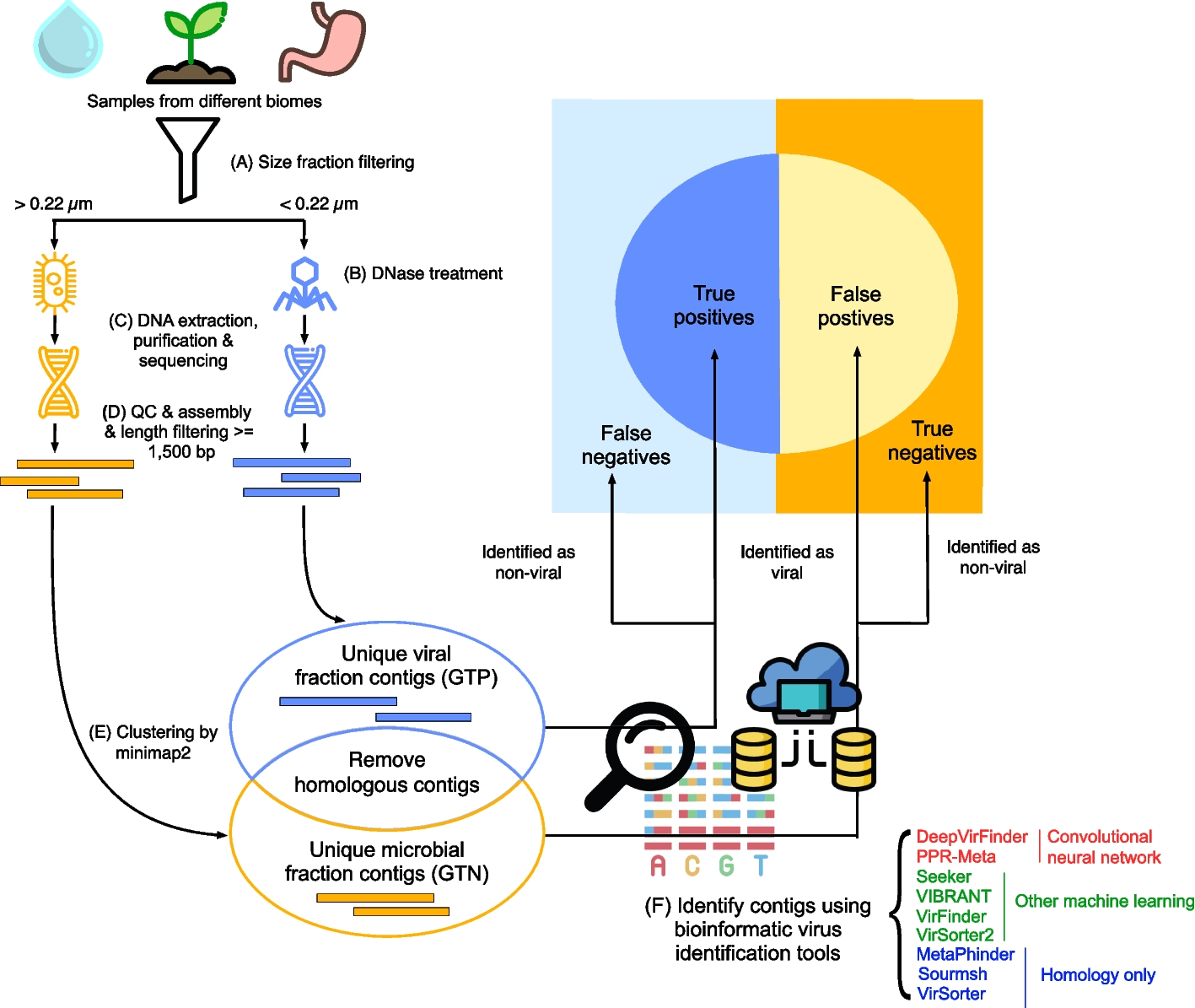




The clust-mst pipeline (see Fig. 1) consists of four parts: (i) sketch creation, (ii) pairwise genome distance computation, (iii) MST generation, and (iv) cluster generation. The clust-greedy pipeline (see Fig. 2) consists of three parts: (i) sketch creation, (ii) distance computation, and (iii) greedy incremental clustering. Both of them support two types of input: a single genome file or a list of genome files. RabbitFX and kseq are used for efficient sequence parsing of the single genome file or the file list, respectively.
After parsing, k-mers (i.e., substrings of length k) are generated by decomposing the genome sequences and their reverse complements in a sliding-window way. Only canonical k-mers (the smallest hash value between a k-mer and its reverse complement) are used to create sketches. S minimum hash values are chosen to compose a sketch where S denotes the sketch size. Note that the hash function should be uniform and deterministic [9]. Uniformity ensures that hash values converted from the input k-mer set of a genome are mapped evenly across the hash value space, which provides a representative sampling of MinHash sketches. Determinism ensures that the same input k-mer always produces the same hash value. Considering efficiency and the features mentioned above, we use the MurmurHash3 function in RabbitTClust. MurmurHash3 [36] is a popular non-cryptographic hash function that converts k-mers to integers. To improve efficiency, we include a vectorized implementation of MurmurHash3 with SIMD instructions which manipulates multiple k-mers concurrently [37].
Distance computation in clust-mst is used to estimate pairwise genome similarities for each pair of sketches. MST generation is done by using a streaming strategy together with distance computation in parallel, as illustrated in Fig. 3. After the MST is constructed, the final clusters are generated by cutting off edges over the threshold in the MST.
For clust-greedy, the sketches are sorted by genome length in descending order. The sketch corresponding to the longest genome is added to the representative set. Each sketch in this set represents a cluster. For each remaining sketch, we compute the distances between the current incoming sketch and all representative sketches. If the distance between this incoming sketch and representative sketches A is the minimum distance and is less than the threshold, the incoming genome is added to the cluster A. If all distances to representative sketches are over the threshold, the incoming sketch is used as the representative of a new representative sketch set. Clustering is finished after all the remaining sketches are processed.
Sketching and distance measurementWe rely on sketching of k-mers for similarity estimation among input genome sequences based on their Mash distance [10] or AAF distance [33] using resemblance Jaccard or containment coefficients. This reduces the size of the input data set by several orders-of-magnitude (the sketch size for Jaccard coefficient and the mean value of variable sketch sizes for containment coefficient are 1000 k-mers per genome by default).
Consider two genomic sequences \(G_1\) and \(G_2\). The Jaccard Index J for their resemblance can be approximated by \(J(G_1,G_2) \approx J(S(G_1),S(G_2)) = |S(G_1)\cap S(G_2)|/|S(G_1)\cup S(G_2)|\) where \(S(G_1)\) and \(S(G_2)\) are hash value sets of the two subsampled k-mer sketches of \(G_1\) and \(G_2\). Mash [10] proposed Mash distance based on a Poisson distribution of the point mutation rate defined as \(D=-\frac \ln \frac\), where k is the k-mer size and J is Jaccard index. Mash distance correlates well with the ANI as \(D\approx 1 - ANI\).
Fixed-size sketches are suitable for computing the resemblance Jaccard coefficient when the lengths of genomes are roughly equal but not in the case of significantly different sizes. When de-duplicating, we also offer a containment analysis option to find duplicate sequences of different sizes. Compared to resemblance, the variable-size-sketch-based containment method is more suitable for genomes with significantly different sizes. The hash value distributions of fixed-size sketches are different when the genomes are of very different sizes, so the matching rate of hashes in the two sketch sets is much smaller than the containment similarity of original genomes, see Fig. 6b. Containment coefficient of \(G_1\) in \(G_2\) is approximated by \(c\approx |S(G_1)\cap S(G_2)|/|S(G_1)|\), whereby sketch sizes are proportional to the size of the respective genomes [38]. AAF distance is defined as \(d=-\frac\ln c\) where c is the containment coefficient [32]. As is shown in Fig. 6b, the matching rate of minimum hashes in the sketch of the smaller genome is similar to the containment similarity between the two genomes.
Fig. 6
Differences between fixed-size and variable-size MinHash sketches on k-mer sets with similar sizes (a) and very different sizes (b). The pair of shaded circles represent two k-mer sets (set A and B) and the overlapping part represents their intersection. The diagrams below each pair circle are the hash sets converted from the k-mer sets. The space of hashes is \(2^b\) with b-bit hash values, leading to a numerical range of hash values between 0 and \(2^b-1\). The points on horizontal lines denote the hash values from set A and B. Solid points are minimum hash values that compose the sketches, while hollow points are hash values not in sketches. Solid arrows represent matching of hashes between two sketches, while dotted lines represent matching not in sketches. In a, sets A and B have similar size, and sketches are composed of 7 minimum hashes (solid points). A and B can get high resemblance from sketches since the distributions of sketch hashes are similar across space \(2^b\). In b, set B is three times the size of set A and contains A totally. The larger set B thus saturates the space more densely. In (\(b_1\)), sketches are both of fixed size of 7. The match rate (1/7) of minimum hashes in the sketches (solid arrow) is much smaller than the containment similarity (7/7) of the original k-mer sets. In (\(b_2\)), sketch sizes are variable and in proportion to the size of respective k-mer sets. Thus, the sketch size (number of solid points) of A is still 7, while the sketch size of B is now 21. The matching rate of minimum hashes in set A is similar to the containment similarity between A and B
Minimum spanning tree for single-linkage hierarchical clusteringHierarchical clustering requires computation of all pairwise distances. The dimension of the distance matrix is \(\mathcal (N^2)\), where N is the number of genomes. It is unpractical to store the whole matrix in memory for large input datasets. However, the memory footprint of the MST is only linear with respect to the number of genomes which is significantly smaller than the whole distance matrix. Thus, we have designed a parallelized streaming approach for MST generation. Subsequently, the MST is chosen to generate clusters by cutting off the edges whose lengths are over a predefined threshold.
Our streaming method is inspired by the edge-partition-based distributed MST algorithm [39]. The all-to-all distance matrix can be considered as a complete graph. In this complete graph, vertices express genomes, and the weights of edges express their pairwise distances. When the graph is partitioned into several sub-graphs, any edge that does not belong to the MST of a sub-graph containing the edge does not belong to the MST of the original graph [40]. This property guarantees that the MST can be constructed by merging sub-MSTs in streaming fashion, which avoids storing the whole distance matrix in memory. In our implementation, the sub-MSTs are concurrently constructed using multiple threads. As shown in Fig. 3, t rows of the distance matrix compose a sub-graph. P sub-MSTs are generated from P sub-graphs concurrently, where P is the thread number. P sub-MSTs are updated as new pairwise distances are calculated. The final MST is merged from the P sub-MSTs after finishing the whole distance computing.
For runtime consideration, the distance computation and sub-MST generation and updating are implemented in parallel by multiple threads. Only P sub-MSTs and P sub-graphs are stored in memory for MST generation. For N input genomes, the magnitude of sub-graphs and sub-MSTs is \(t \cdot N\) and N, respectively. For P threads, the total memory footprint is of a magnitude of \(\mathcal (P \cdot (t+1) \cdot N)\). The parameter t is used to control the dimension of the sub-graphs which is set to 8 by default. Since P and t are much smaller than N, the total memory footprint is typically linear in the number of genomes.
The time consumption of generating clusters from the MST is comparatively small. Since the MST for a dataset will not change as long as the sketch parameters do not change, we store the MST information into an output file. Clusters with different thresholds can be generated from the stored MST file without re-generating the MST again. Users can run with -f option to use the stored MST file as an input.
Note that the MST-based clustering strategy is equal to the single-linkage hierarchical clustering, which may chain two separated clusters together by the noise point. clust-mst takes into account the local density of each genome vertex [41]. For each vertex, the local density is defined as the number of vertices with a distance under the threshold. For each cluster generated by cutting off the over threshold edges of the MST, in default the vertex, x, is labeled as noise when its local density \(dens(x) < min(Q_1, 2)\), where the \(Q_1\) is the first quartile. clust-mst will then cut the edges with the noise vertices to reduce the impact of chaining two clusters together by noise vertices.
Benchmarking clustering accuracyWe use purity and NMI (Normalized Mutual Information) score [42] to assess the quality of clustering results. The ground truth of bacteria genomes from NCBI Refseq and Genbank databases are the species taxonomy identifier (species-taxid) from the assembly summary report files.
Purity is used to measure the degrees of mixing for each cluster. A purity score of 1.0 means the elements in a predicted cluster are all from the same real class. For the predicted clusters P, the ground truth class G, and the total genome number N, the purity can be computed by Eq. 1. However, purity does not penalize scattered cluster result leading to a purity score of 1.0 if each element serves as a single cluster.
NMI is a normalization of the Mutual Information (MI) score to scale the results between 0 (no mutual information) and 1.0 (perfect correlation). Equation (2) describes the MI of predicted clusters P and ground truth class G to reveal the mutual dependency between P and G, where N denotes the total number of genomes. MI is normalized by the average entropy of P and G to scale the results between 0 and 1. Entropy and NMI are computed as shown in Eqs. (3) and (4). NMI score computation is implemented with scikit-learn [43]. The scripts are publicly available in our repository’s evaluation directory [44].
$$\begin purity\left( P;G \right) = \frac\sum _^\max _\left| p \cap g \right| \end$$
(1)
$$\begin MI\left( P;G \right) =\sum _^\ \sum _^\frac\,log\left( \frac \right) \end$$
(2)
$$\begin H\left( X \right) = - \sum _^ \frac\ \log \left( \frac\right) \end$$
(3)
$$\begin NMI\left( P;G \right) = \frac \end$$
(4)
Parameters k-mer sizeConsidering accuracy, the value of k is a trade-off between sensitivity and specificity [10]. Similarities are more sensitive to smaller k since k-mers are more likely to match. However, there is also a higher chance that collisions may inflate the proportion of shared k-mers when the genome size is large. The probability of a specific k-mer appearing in a random nucleotide string of size L is \(1-(1-|\Sigma |^)^\), where the \(\Sigma\) is the alphabet set (\(\Sigma = \)). The k-mer size should be large enough to avoid too many collisions by chance. On the other hand, since k consecutive k-mers will be affected by a single mutation nucleotide, as k grows, the number of matching k-mers between genomes is reduced, leading to lower similarity sensitivity between genomes. The optimal k-mer size needs to be large enough to significantly reduce the chance collisions without losing the similarity sensitivity [33].
To consider both specificity and sensitivity, RabbitTClust firstly scans all input genomes to identify the largest genome length \(L_l\) and average genome length \(L_a\). The k-mer size k can be computed by Eq. 5, where the L is the genome length, and the q is the desired probability of observing a random k-mer [31]. The optimal k-mer size \(k_o\) is computed by Eq. 5 using \(q = 0.0001\) and \(L = L_l\). Furthermore, to protect the accuracy from the chance collisions of k-mers, we define a warning k-mer size \(k_w\) as the lower bound when choosing k. \(k_w\) is computed using \(q = 0.001\) and \(L = L_l\) by Eq. 5. Users can set a specific k-mer size by the option of \(-k\), and when the k-mer size is smaller than \(k_w\), the k-mer size will be reset to the \(k_o\).
$$\begin k=\big \lceil \log _\left( L\left( 1-q \right) /q\right) \big \rceil \end$$
(5)
Sketch sizeMinHash [38] is a fast method to estimate the Jaccard similarity of two sets. It has been proven effective in large-scale genome distance estimation [10]. Thus, we also adopted this method when designing RabbitTClust. The sketch size is the number of minimum hash values in the MinHash sketch. The memory footprint of the sketch for each genome is about 8KB when the sketch size is set to 1000 (each hash value is saved as an unsigned 64-bit integer). In addition to the memory footprint, the sketch size also influences runtime and accuracy. The MinHash algorithm in RabbitTClust maintains a minHash-heap with complexity \(\mathcal (L \cdot log|S|)\) where |S| is sketch size, and L is the genome length. Furthermore, the time of distance computation is linear with respect to the sketch size since it is based on computing the intersections of sets.
The distance estimation accuracy of a pair of genomes improves when using a larger sketch size. Choosing the sketch size is thus a trade-off between the accuracy of distance estimation and efficiency. The memory footprint of sketches and the run time of computing Jaccard or containment are linear in terms of the sketch size. Error bounds decrease with relation to an exponential of the sketch size [30, 45]. For the distance accuracy, as the initial growing stage of sketch size, the distance estimation accuracy increases a lot. But as the sketch size grows further, the distance estimation accuracy increases slightly. RabbitTClust uses a sketch size of 1000 by default, which is a good balance between accuracy and efficiency in most cases.
The sketch size is in proportion with the genome length for the variable-sized sketch of containment coefficient. The variable sketch size is computed as \(|S| = L \cdot P\), where L is the specific genome length and P is the sample proportion. The default sampling proportion \(P_d\) is computed as \(P_d = 1000 / L_a\), where the \(L_a\) is the average genome length (average sketch size is 1000). The sketch size and sample proportion can also be specified by -s and -c options, respectively.
Distance thresholdFor clust-mst, the time consumption of generating clusters from the MST is negligible; i.e., its runtime changes little with different thresholds. However, for clust-greedy the distance threshold dramatically influences the number of pairwise distance computations; i.e., a smaller distance threshold will generate fewer redundant genomes and more clusters. More clusters in turn results in more pairwise distances computation. Thus, runtime of clust-greedy increases with a lower distance threshold.
The MinHash algorithm is a kind of LSH (Locality Sensitive Hashing) with high estimation accuracy for highly similar elements but is less accurate for dissimilar elements [30]. To achieve high cluster accuracy, the distance threshold cannot be too large. With the default sketch size of 1000, the recommended distance threshold should be less than 0.1 with an acceptable distance estimated error. A larger sketch size should be used for higher distance thresholds.
For clustering at the species rank of prokaryotic genomes, the 0.05 distance threshold is used as recommended in Mash and fastANI. Our evaluation shows that clust-mst achieves the best performance on the Bacteria dataset with a distance threshold around of 0.05 (see Fig. 7).
Fig. 7
Evaluation of distance threshold with different sketch sizes for clust-mst on bact-RefSeq dataset (s100 in the figure means the sketch size is 100)
For clust-greedy, the distance measurement is AAF distance corresponding to containment similarity. Different thresholds correspond to different degrees of redundancy. Users can choose different thresholds to filter out various degrees of redundancy by the option of -d.
RabbitTClust determines a valid range of distance thresholds by the k-mer size accompanied with sketch size (for Mash distance) or sampling proportion (for AAF distance). For Mash distance, consider the k-mer size k and sketch size S. The minimum scale interval of Jaccard index is computed by \(J_ = 1 / S\), and the maximum distance threshold is determined by \(d_ = -\frac \ln \frac}}\). For AAF distance, consider the k-mer size k, minimum genome length \(L_\) and sampling proportion P. For variable sketch sizes, the upper bound of the minimum scale interval of containment coefficient is computed as \(C_ = 1 / (L_ \cdot P)\). The maximum recommended distance threshold is then determined by \(d_= -\frac \ln C_\). The chosen distance threshold should be less than the \(d_\) in order to generate the clusters in a valid range.
留言 (0)