
記住我
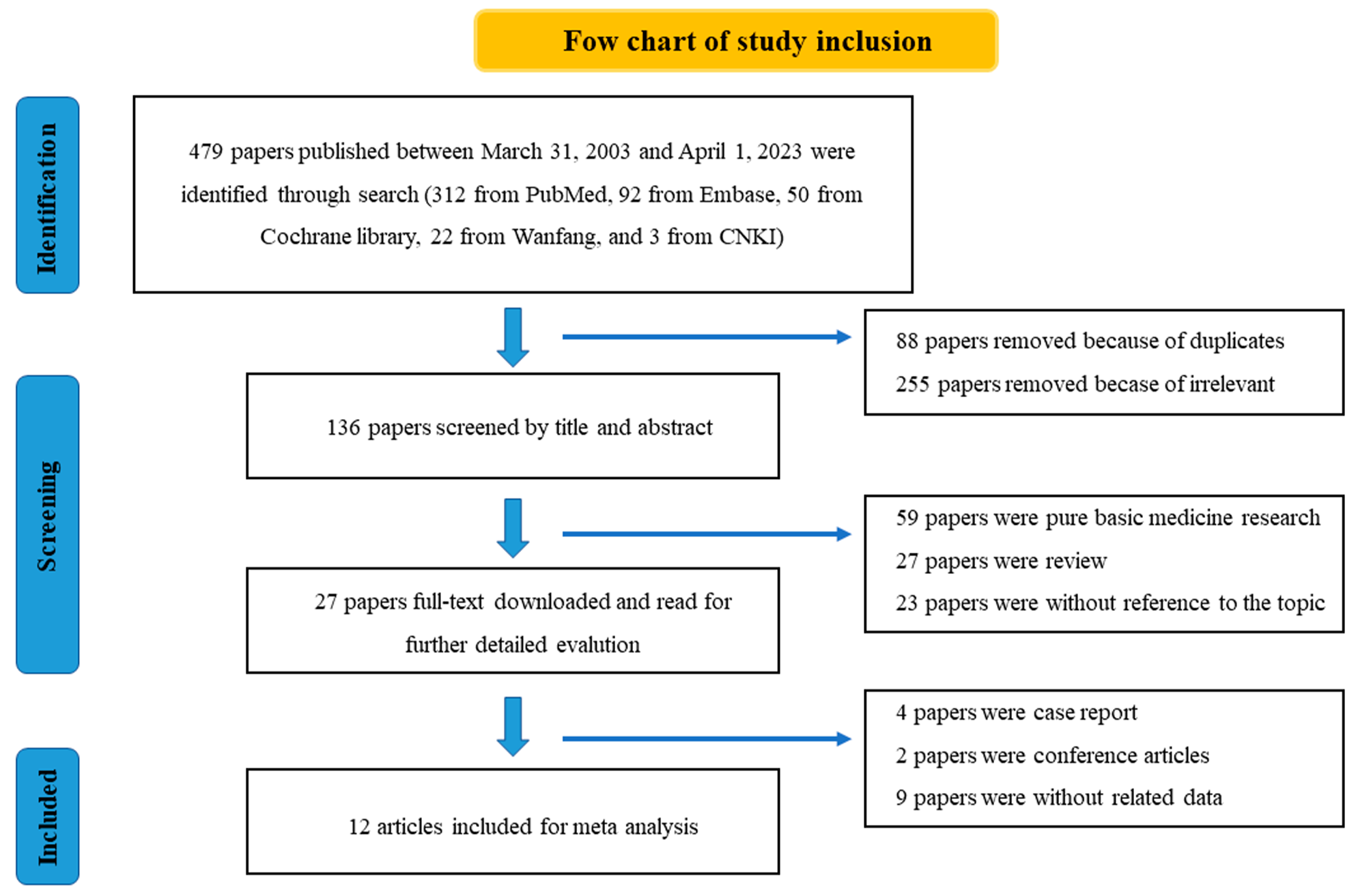

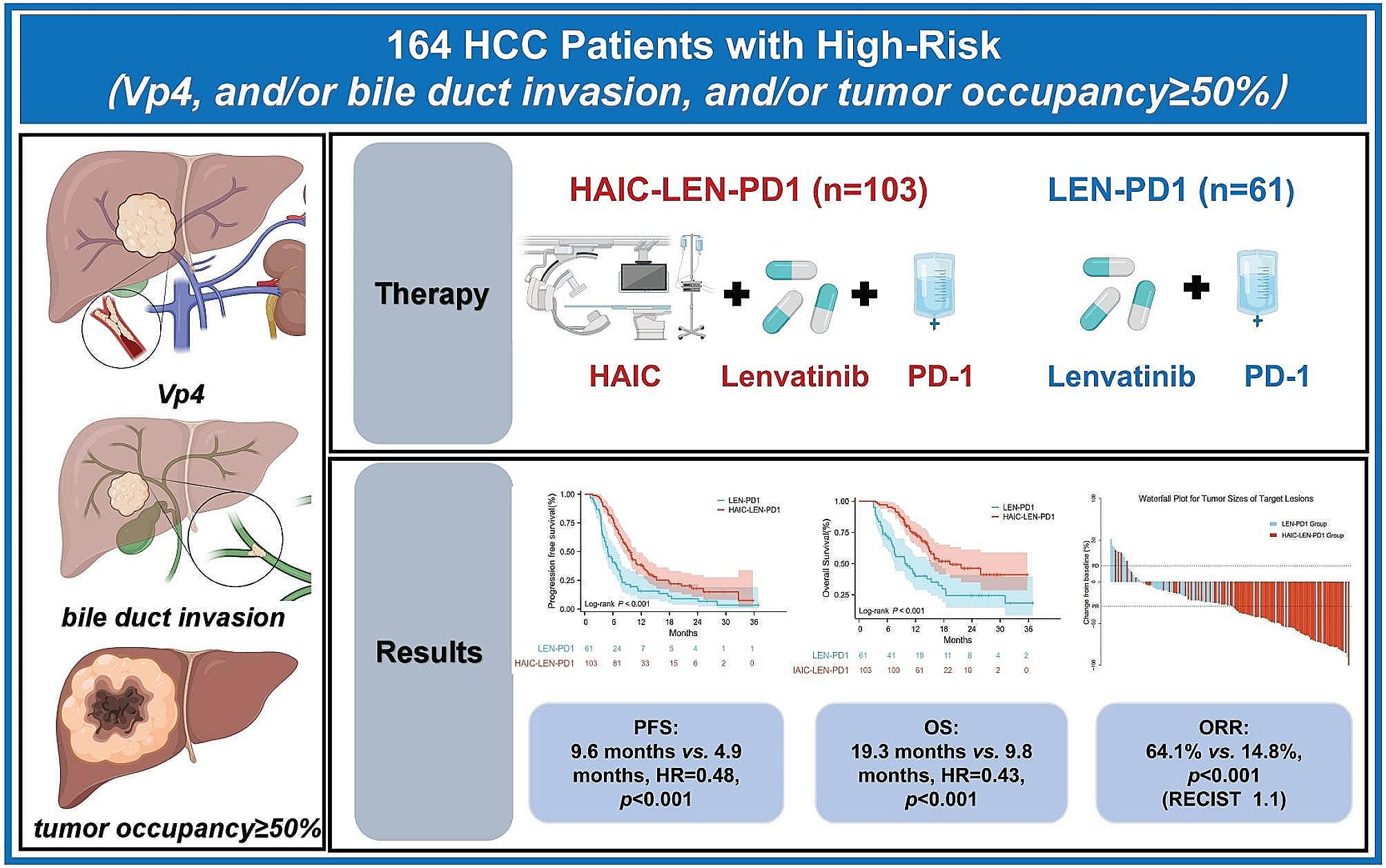
In this cross-sectional analysis, we found that post-progression treatment data were not available in 60% of publications and 49% of registration trials. Among trials with assessable post-progression data, the treatment was substandard in 55% of published trials and 76% of registration trials. We conducted a subgroup analysis in trials reporting statistically significant positive OS results and found that the proportion of post-progression therapy reporting was higher than in the overall cohort. Yet, when assessable, post-progression was appropriate in fewer trials than in the overall cohort.
Our findings show deficiency in the level of reporting and quality of subsequent treatment that patients may receive when enrolled in oncology RCTs. Similar findings have previously been reported in specific tumor types like in multiple myeloma [4] or renal cell carcinoma [5]. The need for conducting trials globally, which include middle and low-income countries with limited or no access to SOC, is one explanation for suboptimal post-progression treatment. However, this is not acceptable in our view, as these same trials are often the basis on which approvals, guidelines, and recommendations are implemented in high-income countries.
Post-progression treatment access is indeed critical in appraising the results of RCT’s since substandard post-progression treatment may limit the generalizability of the reported results to places where optimal care is available. In line with a previous work [3], we identified three situations when undesirable post-progression treatment may occur (Fig. 4).
Fig. 4
« Ten percent» pre-specified rules to assess post-progression treatment in the control arm of randomized clinical trials. Legend: post-progression therapy is referring to systemic treatment
First, when a treatment is already approved in subsequent lines and tested upfront, the core question of the trial is if there is benefit in moving the drug upfront. Free access to the treatment at progression in the control arm should therefore be mandatory. If not, the question whether upfront treatment is superior to the current standard, being treatment when the disease progresses, will remain unanswered (Fig S1). The LATITUDE trial randomized patients with metastatic castration-sensitive prostate cancer between androgen deprivation therapy alone or with abiraterone [10, 16]. In castration-resistant patients, abiraterone was SOC before the trial started enrolment. However, in LATITUDE, only 24% of control arm patients treated with a subsequent therapy received abiraterone [10]. As we previously highlight, “we cannot be sure that the survival advantage of early treatment would still exist if control patients had fair access to this drug” [1, 3].
Second, inappropriate use of crossover may confound a true verdict on the drug’s therapeutic effect. If a novel treatment is tested (with unproven efficacy in subsequent lines), patients in the control arm should not be offered this treatment at progression, and should not crossover. If the drug truly has a survival gain, crossover may obscure the benefit. Conversely, if the drug offers no survival gain, crossover may lead to a delay in the time when effective therapies will be provided. This may lead to superior OS in the experimental arm (not subject to crossover) being incorrectly imputed to better efficacy when it may be merely due to the fact that the intervention arm received superior post-progression therapy. The IMPACT study trial tested sipuleucel-T in patients with metastatic castration-resistant prostate cancer [17]. The crossover to sipuleucel-T in the control arm may have delayed access and lowered the proportion of patients in the control arm having access to life-prolonging therapies, possibly explaining the OS benefit with no PFS advantage (Fig S2) [3]. For these reasons crossover is not desirable in trials assessing the fundamental efficacy of products, and is discouraged by regulatory agencies [3, 18,19,20].
The last scenario is when the proportion of patients receiving post-progression treatment in the trial is inferior to real-world settings. Because of strict inclusion–exclusion criteria, trials are known to select patients with better performance status and fewer comorbidities [7]. It has been shown that 38% of patients in the real-world would be ineligible for trial participation [8]. As a consequence, because trial patients are generally healthier and more fit than average cancer patients, the proportion of patients receiving a subsequent therapy in trials should be higher than in real-life. If not, this limitation will affect both arms. However, we contend, as others do, that such a trial may favor the experimental arm and be more likely to conclude a survival benefit when the real-world use of the drug would not yield a similar result. Many researchers state that post-progression treatment can “dilute” the PFS or overall response rate advantage [21, 22]. The MONALEESA-7 trial tested ribociclib against placebo (plus hormonal therapy) in patients mostly in the first-line setting of hormone-sensitive advanced or metastatic breast cancer [23]. Post-progression treatment was low in both arms (73% in the control arm, 69% in the experimental arm) [14], when real-life data demonstrated higher access (up to 92% after first-line hormonal therapy) [24]. Would the same OS advantage have occurred if both arms had optimal access to subsequent therapeutic options? The dilution is precisely the question at hand: there is no reason to give the drug sooner, with the potential of more toxicity, if subsequent therapies achieve the same result (Fig S3).
Examples of desirable and undesirable scenarios are provided in Table 2. We also described the “ten percent” rules, which we prespecified and applied to assess post-progression treatment in our work (Fig. 4). Others may disagree, and they can propose and apply other rules, and we encourage this effort.
Assessment scores for the value of anti-cancer treatments, such as the ASCO-Value Framework Net Health Benefit and ESMO-MCBS, do not consider the quality of post-progression therapy. Similarly, the Cochrane Risk of Bias assessment (RoB2) does not systematically evaluate post-progression data. Yet, when analyzing results from clinical trials that have suboptimal post-progression therapy, it is crucial to be aware of these limitations in order to make informed clinical decisions. For instance, a trial with proper post-progression care will likely produce more trustworthy results compared to a trial with inadequate or unreported post-progression data. Ultimately, the lack of trials that accurately reflect standard practice is a significant issue. One potential solution to this problem is for regulatory bodies to only grant marketing authorization for trials that provide optimal post-progression therapy in both the experimental and control groups.
LimitationsOur work has strengths and limitations. First, the time-period is limited to three years, and this is not a systematic review. Our aim was to capture recent trends in reporting post-progression therapy, and we encourage others to expand upon our work to cover a broader time period. However, our work is the first to encompass all tumor-types, and to conduct analyses in both FDA registration trials and top journal publications. Second, the prespecified rules can be questioned and are not supported by randomized data. We prespecified them to allow for reproducibility, and they were based on previous work of our group. Also, we detailed the proportion of trials coded with each rule to show their relative prevalence. Last, in ambiguous cases, trials were considered appropriate, so our work may have overestimated the proportion of trials with optimal post-progression treatment.
留言 (0)